57 Harmful effects of drugs
Overview
This chapter addresses harmful effects of drugs, both in the context of therapeutic use—so-called adverse drug reactions, and of deliberate overdose. The classification of adverse drug reactions is considered, followed by aspects of drug toxicity: toxicity testing in drug development, mechanisms of toxin-induced cell damage, mutagenesis and carcinogenicity, teratogenesis and allergic reactions.
Introduction
Paracelsus, a 16th-century alchemist, is credited with the aphorism that all drugs are poisons: ‘… the dosage makes it either a poison or a remedy’. Today, toxic effects of drugs remain clinically important in the context of deliberate overdose (self-poisoning accounts for approximately 10% of the workload of emergency medicine departments in the UK; by contrast, homicidal poisoning, while obviously important, is extremely uncommon). Some susceptible individuals may experience dose-related toxicity even during therapeutic dosing; some of this susceptibility is genetically determined, and genomic testing as a means of avoiding such harms is beginning to make its way into the clinic (Ch. 11).
Rigorous toxicity testing in animals (see below), including tests for carcinogenicity, teratogenicity and organ-specific toxicities, is carried out on potential new drugs during development (see Ch. 60), and in many cases leads to abandonment of the compound before it is tested in humans. Such animal toxicity studies form part of the package of information routinely submitted to drug regulatory agencies when seeking approval to market a new drug. Such studies do sometimes usefully focus attention on a particular organ, the function of which can be monitored prospectively during human studies. Nevertheless, harmful effects are often encountered during therapeutic use, often the result of misprescribing, but also due to the emergence of toxic effects not detected in animals. These harms are usually referred to as ‘adverse drug reactions’ (ADRs) and are of great concern to drug regulatory authorities, which are charged with establishing the safety as well as the efficacy of drugs. Unpredictable events are of particular concern. Some ADRs are a consequence of the main pharmacological effect of the drug but some (e.g. immunological reactions), are not. Safety (as distinct from toxicity) of new drugs can only be established during drug development and therapeutic use in humans (Walker, 2004).
Clinically important ADRs are common, costly and avoidable (see Pirmohamed et al., 2004).1 Any organ can be the principal target, and several systems can be involved simultaneously. The time course helps to recognise a clinical event as an ADR. Several patterns are recognised. The symptoms sometimes closely shadow drug administration and discontinuation, but in other cases adverse effects only occur during prolonged use (osteoporosis during continued high-dose glucocorticoid therapy [Ch. 32], or tardive dyskinesia during continuous use of antipsychotic drugs [Ch. 45], for example). Some adverse effects occur on ending treatment, either within a few days (e.g. tachycardia on abrupt discontinuation of β-adrenoceptor blockade) or after a delay, first appearing months or years after treatment is discontinued, as in the case of some second malignancies following successful chemotherapy. Consequently, anticipating, avoiding, recognising and responding to adverse drug reactions are among the most challenging and important parts of clinical practice.
Classification of Adverse Drug Reactions
Harmful effects of drugs are either related or unrelated to the principal pharmacological action of the drug. Aronson & Ferner (2003) have suggested that ADRs be described according to the dose, time course and susceptibility (DoTS).
Adverse Effects Related to the Main Pharmacological Action of the Drug
Many adverse effects related to the main pharmacological action of the drug are predictable, at least if this action is well understood. They are sometimes referred to as type A (‘augmented’) adverse reactions (Rawlins & Thomson, 1985) and are related to dose and susceptibility. Many such reactions have been described in previous chapters. For example, postural hypotension occurs with α1-adrenoceptor antagonists, bleeding with anticoagulants, sedation with anxiolytics and so on. In many instances, this type of unwanted effect is reversible, and the problem can often be dealt with by reducing the dose. Such effects are sometimes serious (e.g. intracerebral bleeding caused by anticoagulants, hypoglycaemic coma from insulin), and occasionally they are not easily reversible, for example drug dependence produced by opioid analgesics (see Ch. 48).
Some adverse effects related to the main action of a drug result in discrete events rather than graded symptoms, and can be difficult to detect. For example, drugs that block cyclo-oxygenase (COX)-2 (including ‘coxibs’, for example rofecoxib, celecoxib, valdecoxib, as well as some conventional non-steroidal anti-inflammatory drugs, NSAIDs) increase the risk of myocardial infarction in a dose-dependent manner (Ch. 26). This potential was apparent from the pharmacology of these drugs, in particular their ability to inhibit prostacyclin biosynthesis as well as to increase arterial blood pressure, and early studies gave a hint of such problems. The effect was difficult to prove because of the high background incidence of coronary thrombosis, and it was only when placebo-controlled trials were performed for another indication (in the hope that COX-2 inhibitors could prevent bowel cancer) that this effect was confirmed unequivocally.
Adverse Effects Unrelated to the Main Pharmacological Action of the Drug
Adverse effects unrelated to the main pharmacological effect may be predictable when a drug is taken in excessive dose, for example paracetamol hepatotoxicity (see below) or aspirin-induced tinnitus; or when susceptibility is increased, for example during pregnancy or by a predisposing disorder such as glucose 6-phosphate dehydrogenase deficiency or a mutation in the mitochondrial DNA that predisposes to aminoglycoside ototoxicity (Ch. 11).
Unpredictable idiosyncratic reactions are often initiated by a chemically reactive metabolite rather than the parent drug. Examples of such ADRs, which are often immunological in nature, include drug-induced hepatic or renal necrosis, bone marrow suppression, carcinogenesis and disordered fetal development. Uncommon but severe unpredictable adverse effects that have been mentioned in earlier chapters include aplastic anaemia from chloramphenicol and anaphylaxis in response to penicillin. These idiosyncratic reactions are termed type B (‘bizarre’) in the Rawlins & Thomson (1985) classification. They are usually severe—otherwise they would go unrecognised—and their existence is important in establishing the safety of medicines.
 If the incidence of an adverse reaction is 1 in 6000 patients exposed, approximately 18 000 patients would have to be exposed to the drug for three events to occur, and approximately double that number for three events to be detected and their possible relationship to the drug recognised and reported, even if there were no background incidence of the event in question. Consequently, such reactions cannot be excluded by preapproval clinical trials (which might typically expose only a few thousand individuals to the drug), and the association may come to light only after years of use, so there is a need for continued monitoring by regulatory authorities after drugs have been licensed and marketed. An example is the association between pulmonary hypertension and valvular heart disease with fenfluramine, an appetite suppressant that had been used for several years, and with dexfenfluramine, its pharmacologically active isomer. Such experiences call for a balanced approach to prescribing new drugs if there are adequate existing alternatives.2 This conflicts with the culture of drug marketing, especially when this involves advertising the product direct to the consumer.
If the incidence of an adverse reaction is 1 in 6000 patients exposed, approximately 18 000 patients would have to be exposed to the drug for three events to occur, and approximately double that number for three events to be detected and their possible relationship to the drug recognised and reported, even if there were no background incidence of the event in question. Consequently, such reactions cannot be excluded by preapproval clinical trials (which might typically expose only a few thousand individuals to the drug), and the association may come to light only after years of use, so there is a need for continued monitoring by regulatory authorities after drugs have been licensed and marketed. An example is the association between pulmonary hypertension and valvular heart disease with fenfluramine, an appetite suppressant that had been used for several years, and with dexfenfluramine, its pharmacologically active isomer. Such experiences call for a balanced approach to prescribing new drugs if there are adequate existing alternatives.2 This conflicts with the culture of drug marketing, especially when this involves advertising the product direct to the consumer.
Drug Toxicity
Toxicity Testing
Toxicity testing in animals is carried out on new drugs to identify potential hazards before administering them to humans. It involves the use of a wide range of tests in different species, with long-term administration of the drug, regular monitoring for physiological or biochemical abnormalities, and a detailed postmortem examination at the end of the trial to detect any gross or histological abnormalities. Recently, use of non-mammalian species, notably the transparent zebra fish, has shown promise as an intermediate stage between toxicity studies on cells and tissues in vitro and mammalian toxicity testing (see Parng, 2005, for a review). Toxicity testing is performed with doses well above the expected therapeutic range, and establishes which tissues or organs are likely ‘targets’ of toxic effects of the drug. Recovery studies are performed to assess whether toxic effects are reversible, and particular attention is paid to irreversible changes such as carcinogenesis or neurodegeneration. The basic premise is that toxic effects caused by a drug are similar in humans and other animals. This is inherently reasonable in view of the similarities between higher organisms at the cellular and molecular levels. There are, nevertheless, wide interspecies variations, especially in metabolising enzymes; consequently, a toxic metabolite formed in one species may not be formed in another, and so toxicity testing in animals is not always a reliable guide. Pronethalol, the first β-adrenoceptor antagonist synthesised (by James Black) at ICI, was not developed because it caused carcinogenicity in mice; it subsequently emerged that carcinogenicity occurred only in the ICI strain—but by then other β-blockers were already in development.
Toxic effects can range from negligible to so severe as to preclude further development of the compound. Intermediate levels of toxicity are more acceptable in drugs intended for severe illnesses (e.g. AIDS or cancers), and decisions on whether or not to continue development are often difficult. If development does proceed, safety monitoring can be concentrated on the system ‘flagged’ as a potential target of toxicity by the animal studies.3 Safety of a drug (as distinct from toxicity) can be established only during use in humans.
Types of drug toxicity 
General Mechanisms of Toxin-Induced Cell Damage and Cell Death
Toxic concentrations of drugs or drug metabolites can cause necrosis; however, programmed cell death (apoptosis; see Ch. 5) is increasingly recognised to be of paramount importance, especially in chronic toxicity (see, for example, Pirmohamed, 2003).
Chemically reactive drug metabolites can form covalent bonds with target molecules as well as damage tissue by non-covalent mechanisms. The liver is of great importance in drug metabolism (Ch. 9), and hepatocytes are exposed to high concentrations of nascent metabolites. Drugs and their polar metabolites are concentrated in renal tubular fluid as water is reabsorbed, so renal tubules are exposed to higher concentrations than are other tissues. Several hepatotoxic drugs (e.g. paracetamol) are also nephrotoxic. Consequently, hepatic or renal damage are common reasons for abandoning development of drugs during toxicity testing.
Non-Covalent Interactions
Reactive metabolites of drugs are implicated in several potentially cytotoxic, non-covalent processes, including:
Lipid peroxidation
Peroxidation of unsaturated lipids can be initiated either by reactive metabolites or by reactive oxygen species (see below). Lipid peroxyradicals (ROO•) can produce lipid hydroperoxides (ROOH), which produce further lipid peroxyradicals. This chain reaction—a peroxidative cascade—may eventually affect much of the membrane lipid. Defence mechanisms, for example GSH peroxidase and vitamin E, protect against this. Cell damage results from alteration of membrane permeability or from reactions of the products of lipid peroxidation with proteins.
Reactive oxygen species
Reduction of molecular oxygen to superoxide anion (O2−•) may be followed by enzymic conversion to hydrogen peroxide (H2O2), hydroperoxy (HOO•) and hydroxyl (OH•) radicals or singlet oxygen. These reactive oxygen species are cytotoxic, both directly and through lipid peroxidation (see above), and are important in excitotoxicity and neurodegeneration (Ch. 39, Fig. 39.1).
Depletion of glutathione
The GSH redox cycle protects cells from oxidative stress. GSH can be depleted by accumulation of normal oxidative products of cell metabolism, or by the action of toxic chemicals. GSH is normally maintained in a redox couple with its disulfide, GSSG. Oxidising species convert GSH to GSSG, GSH being regenerated by NADPH-dependent GSSG reductase. When cellular GSH falls to about 20–30% of normal, cellular defence against toxic compounds is impaired and cell death can result.
Modification of sulfhydryl groups
Modification of sulfhydryl groups can be produced either by oxidising species that alter sulfhydryl groups reversibly or by covalent interaction. Free sulfhydryl groups have a critical role in the catalytic activity of many enzymes. Important targets for sulfhydryl modification by reactive metabolites include the cytoskeletal protein actin, GSH reductase (see above) and Ca2+-transporting ATPases in the plasma membrane and endoplasmic reticulum. These maintain cytoplasmic Ca2+ concentration at approximately 0.1 µmol/l in the face of an extracellular Ca2+ concentration of more than 1 mmol/l. A sustained rise in cell Ca2+ occurs with inactivation of these enzymes (or with increased membrane permeability; see above), and this compromises cell viability. Lethal processes leading to cell death after acute Ca2+ overload include activation of degradative enzymes (neutral proteases, phospholipases, endonucleases) and protein kinases, mitochondrial damage and cytoskeletal alterations (e.g. modification of association between actin and actin-binding proteins).
Covalent Interactions
Targets for covalent interactions include DNA, proteins/peptides, lipids and carbohydrates. Covalent bonding to DNA is a basic mechanism of mutagenic chemicals; this is dealt with below. Several non-mutagenic chemicals also form covalent bonds with macromolecules, but the relationship between this and cell damage is incompletely understood. For example, the cholinesterase inhibitor paraoxon (the active metabolite of the insecticide parathion) binds acetylcholinesterase at the neuromuscular junction (Ch. 13) and causes necrosis of skeletal muscle. One toxin from an exceptionally poisonous toadstool, Amanita phalloides, binds actin, and another binds RNA polymerase, interfering with actin depolymerisation and protein synthesis, respectively.
General mechanisms of cell damage and cell death
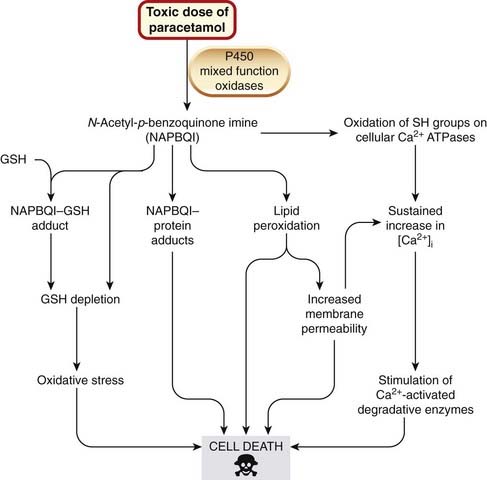
Hepatotoxicity
Many therapeutic drugs cause liver damage, manifested clinically as hepatitis or (in less severe cases) only as laboratory abnormalities (e.g. increased activity of plasma aspartate transaminase, an enzyme released from damaged liver cells). Paracetamol, iproniazid and halothane cause hepatotoxicity by the mechanisms of cell damage outlined above. Genetic differences in drug metabolism (see Ch. 11) have been implicated in some instances (e.g. isoniazid, phenytoin). Mild drug-induced abnormalities of liver function are not uncommon, but the mechanism of liver injury is often uncertain (e.g. statins; Ch. 23). It is not always necessary to discontinue a drug when such mild laboratory abnormalities occur, but the occurrence of cirrhosis as a result of long-term low-dose methotrexate treatment for arthritis or psoriasis (a chronic scaling skin disease of unknown cause that is usually mild, if tiresome, but can rarely be very severe4) argues for caution. Hepatotoxicity of a different kind, namely reversible obstructive jaundice, occurs with chlorpromazine (Ch. 45) and androgens (Ch. 34).
Hepatotoxicity caused by paracetamol overdose remains a common cause of death following self-poisoning. An outline is given in Chapter 26. Because the body’s handling of this drug exemplifies many of the general mechanisms of cell damage outlined above, the story is taken up again here. With toxic doses of paracetamol, the enzymes catalysing the normal conjugation reactions are saturated, and mixed-function oxidases convert the drug to the reactive metabolite N-acetyl-p-benzoquinone imine (NAPBQI). As explained in Chapters 9 and 56, paracetamol toxicity is increased in patients in whom P450 enzymes have been induced, for instance by chronic excessive consumption of alcohol. NAPBQI initiates several of the covalent and non-covalent interactions described above and illustrated in Figure 57.1. Oxidative stress from GSH depletion is important in leading to cell death. Regeneration of GSH from GSSG depends on the availability of cysteine, the intracellular availability of which can be limiting. Acetylcysteine or methionine can substitute for cysteine, increasing GSH availability and reducing mortality in patients with paracetamol poisoning.
Liver damage can also be produced by immunological mechanisms (see below), which have been particularly implicated in halothane hepatitis (see Ch. 40).
Hepatotoxicity
Nephrotoxicity
Drug-induced nephrotoxicity is a common clinical problem: NSAIDs (Table 57.1) and angiotensin-converting enzyme (ACE) inhibitors are among the commoner precipitants of acute renal failure. This is usually caused by the principal pharmacological actions of these drugs, which, although well tolerated in healthy people, cause renal failure in patients with diseases that jeopardise glomerular filtration. In patients with heart or liver disease, glomerular filtration rate (GFR) depends critically on vasodilator prostaglandin biosynthesis. This is inhibited by NSAIDs (Ch. 26), and hence these drugs reduce renal perfusion in such patients. Similarly, in patients with bilateral renal artery stenosis (i.e. narrowings of the renal arteries, most often caused by fibromuscular tissue in young women or by atheromatous disease in older people), GFR depends on angiotensin II-mediated efferent arteriolar vasoconstriction (which is inhibited by ACE inhibitors; Ch. 22); acute renal impairment occurs on starting treatment with an ACE inhibitor and is reversible if the drug is discontinued promptly. Additionally, NSAIDs indirectly depress renin and aldosterone secretion by inhibiting renal prostaglandin I2 biosynthesis, and ACE inhibitors depress angiotensin II-stimulated aldosterone secretion, leading to low renin/low aldosterone states (‘hyporeninaemic hypoaldosteronism’) that are particularly notable in diabetic patients. Reduced aldosterone can cause hyperkalaemia, especially if GFR is also reduced.
Table 57.1 Adverse effects of non-steroidal anti-inflammatory drugs on the kidney
| Cause | Adverse effects |
|---|---|
| Principal pharmacological action (i.e. inhibition of prostaglandin biosynthesis) | Acute ischaemic renal failure Sodium retention (leading to or exacerbating hypertension and/or heart failure) Water retention Hyporeninaemic hypoaldosteronism (leading to hyperkalaemia) |
| Unrelated to principal pharmacological action (allergic-type interstitial nephritis) | Renal failure Proteinuria |
| Unknown whether or not related to principal pharmacological action (analgesic nephropathy) | Papillary necrosis Chronic renal failure |
Adapted from Murray & Brater 1993.
In addition to effects related to their main pharmacological action, NSAIDs can also cause interstitial nephritis through an immunological mechanism. This presents several months to 1 year after starting treatment as acute renal failure, often accompanied by eosinophil leukocytes in the urine and proteinuria, or as nephrotic syndrome (heavy proteinuria, hypoalbuminuria and oedema). Fenoprofen is particularly liable to cause this type of renal damage, possibly because its metabolites bind irreversibly to albumin. Penicillins (Ch. 50), especially meticillin, also cause interstitial nephritis.
Analgesic nephropathy is a third kind of renal damage in which NSAIDs are implicated (see also Ch. 26). This consists of renal papillary necrosis5 and chronic interstitial nephritis. The clinical course is typically insidious but leads ultimately to end-stage chronic renal failure. It is associated with prolonged and massive overuse of analgesics. Phenacetin has been incriminated, but other NSAIDs have not been exonerated. The role of caffeine (often included with analgesics and NSAIDs in combined preparations for migraine) is uncertain but could be important. It is possible that such analgesic-associated nephropathy is causally related to inhibition of renal prostaglandin synthesis, but its pathogenesis is not understood.
Captopril, in higher doses than are currently recommended, can cause heavy proteinuria (Ch. 22). This is the result of glomerular injury, which is also caused by some other drugs that, like captopril, contain a sulfhydryl group (e.g. penicillamine; Ch. 26). It is therefore believed that it is this chemical feature rather than ACE inhibition per se that is responsible for this adverse effect.
Ciclosporin, used to prevent transplant rejection (Ch. 26), causes renal damage via renal vasoconstriction, which reduces GFR and causes hypertension.
Mutagenesis and Carcinogenicity
Chemical agents cause mutation by covalent modification of DNA. Certain kinds of mutation result in carcinogenesis, because the affected DNA sequence codes for a protein that regulates cell growth. It usually requires more than one mutation in a cell to initiate the changes that result in malignancy, mutations in proto-oncogenes (which regulate cell growth) and tumour suppressor genes (which code for products that inhibit the transcription of oncogenes) being particularly implicated (see Chs 5 and 55).
Biochemical Mechanisms of Mutagenesis
Most chemical carcinogens act by modifying bases in DNA, particularly guanine, the O6 and N7 positions of which readily combine covalently with reactive metabolites of chemical carcinogens. Substitution at the O6 position is the more likely to produce a permanent mutagenic effect, because N7 substitutions are usually quickly repaired.
The accessibility of bases in DNA to chemical attack is greatest when DNA is in the process of replication (i.e. during cell division). The likelihood of genetic damage by many mutagens is therefore related to the frequency of cell division. The developing fetus is particularly susceptible, and mutagens are also potentially teratogenic (see below). This is also important in relation to mutagenesis of germ cells, particularly in girls, because in humans the production of primary oocytes occurs by a rapid succession of mitotic divisions very early in embryogenesis. Each primary oocyte then undergoes only two further divisions much later in life, at the time of ovulation. It is consequently during early pregnancy that germ cells of the developing female embryo are most likely to undergo mutagenesis, the mutations being transmitted to progeny conceived many years after exposure to the mutagen. In the male, germ cell divisions occur throughout life, and sensitivity of germ cells to mutagens is continuously present.
Carcinogenesis
Alteration of DNA is the first step in the complex, multistage process of carcinogenesis (see Ch. 5). Carcinogens are chemical substances that cause cancer, and can interact directly with DNA (genotoxic carcinogens) or act at a later stage to increase the likelihood that mutation will result in a tumour (epigenetic carcinogens; Fig. 57.2).
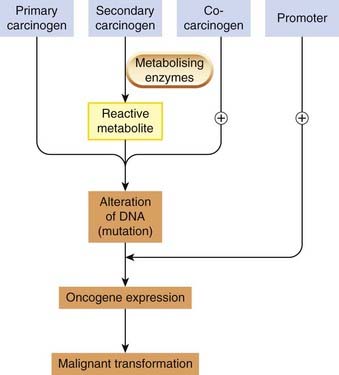
Measurement of Mutagenicity and Carcinogenicity
Much effort has gone into developing assays to detect mutagenicity and carcinogenicity. In vitro tests for mutagenicity are used for screening large numbers of compounds but as predictors of carcinogenicity can give false positive or false negative results. Whole-animal tests for carcinogenicity tests are expensive and time-consuming but are usually required by regulatory authorities before a new drug is licensed for use in humans. The main limitation of this kind of study is that there are important species differences, mainly to do with the metabolism of the foreign compound and the formation of reactive products.
The most widely used in vitro tests are variations on the Ames test for mutagenicity, which measures the rate of back-mutation (i.e. reversion from mutant to wild-type form) in Salmonella typhimurium.
The wild-type strain can grow in a medium containing no added amino acids, because it can synthesise all the amino acids it needs from simple carbon and nitrogen sources. The test makes use of the fact that a mutant form of the organism cannot make histidine in this way and therefore grows only on a medium containing this amino acid. The test involves growing the mutant form on a medium containing a small amount of histidine, the drug to be tested being added to the culture. After several divisions, the histidine becomes depleted, and the only cells that continue dividing are those that have back-mutated to the wild type. A count of colonies following subculture on plates deficient in histidine gives a measure of the mutation rate.
Primary carcinogens cause mutation by a direct action on bacterial DNA, but most carcinogens have to be converted to an active metabolite (see above). Therefore it is necessary to include, in the culture, enzymes that catalyse the necessary conversion. An extract of liver from a rat treated with phenobarbital to induce liver enzymes is usually employed. There are many variations based on the same principle.
Other short-term in vitro tests for genotoxic chemicals include measurements of mutagenesis in mouse lymphoma cells, and assays for chromosome aberrations and sister chromatid exchanges in Chinese hamster ovary cells. However, all the in vitro tests give some false positive and some false negative results.
In vivo tests for carcinogenicity entail detection of tumours in groups of test animals. Carcinogenicity tests are inevitably slow, because there is usually a latency of months or years before tumours develop. Furthermore, tumours can develop spontaneously in control animals, and the results often provide only equivocal evidence of carcinogenicity of the test drug, making it difficult for industry and regulatory authorities to decide on further development and possible licensing of a product. None of the tests so far described can reliably detect epigenetic carcinogens. To do this, it is necessary to measure the effect of the test substance on tumour production with a threshold dose of a genotoxic agent. Such tests are being evaluated.
Few therapeutic drugs are known to increase the risk of cancer, the most important groups being drugs that act on DNA, i.e. cytotoxic and immunosuppressant drugs (Chs 55 and 26, respectively), and sex hormones (e.g. oestrogens, Ch. 34). Pyrimethamine (Ch. 53) is mutagenic in high concentrations, and carcinogenicity testing in strain A mice (but not other strains or species) was positive for a three-fold increase in lung tumours. Methoxsalen (a psoralen used together with ultraviolet light, PUVA, in specialist skin disease centres for treatment of psoriasis) is both mutagenic and carcinogenic in animal models and may increase the incidence of skin cancer in humans.
Carcinogens
Teratogenesis and Drug-Induced Fetal Damage
Teratogenesis signifies the production of gross structural malformations during fetal development, in distinction from other kinds of drug-induced fetal damage such as growth retardation, dysplasia (e.g. iodide-associated goitre) or the asymmetrical limb reduction resulting from vasoconstriction caused by cocaine (see Ch. 48) in an otherwise normally developing limb. Examples of drugs that affect fetal development adversely are given in Table 57.2.
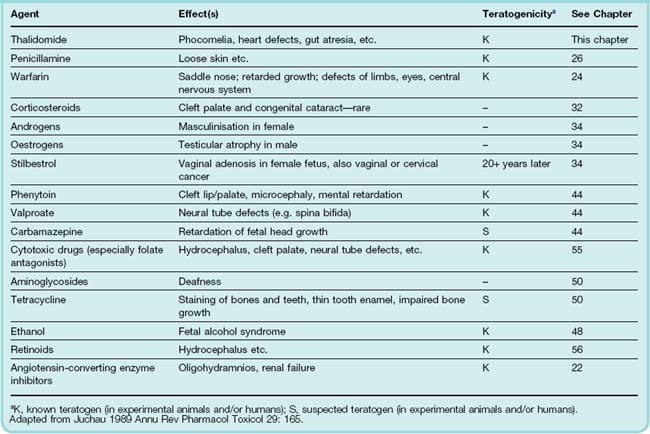
It has been known that external agents can affect fetal development since the 1920s, when it was discovered that X irradiation during pregnancy causes fetal malformation. The importance of rubella infection was recognised two decades later, but it was not until 1960 that drugs were implicated as causative agents in teratogenesis: the shocking experience with thalidomide led to a widespread reappraisal of many other drugs in clinical use, and to the setting up of drug regulatory bodies in many countries. Most birth defects (about 70%) occur with no recognisable causative factor. Drug or chemical exposure during pregnancy is estimated to account for only approximately 1% of all fetal malformations. Fetal malformations are common, so the absolute numbers of children affected are substantial.
Mechanism of Teratogenesis
The timing of the teratogenic insult in relation to fetal development is critical in determining the type and extent of damage. Mammalian fetal development passes through three phases (Table 57.3):
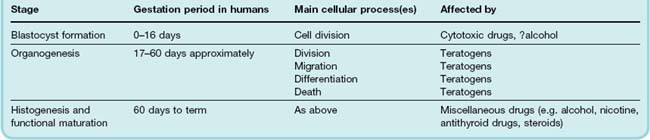
Cell division is the main process occurring during blastocyst formation. During this phase, drugs can kill the embryo by inhibiting cell division, but provided the embryo survives, its subsequent development does not generally seem to be compromised. Ethanol is an exception, affecting development at this very early stage (Ch. 48).
Drugs can cause gross malformations if administered during organogenesis (days 17–60 in humans). The structural organisation of the embryo occurs in a well-defined sequence: eye and brain, skeleton and limbs, heart and major vessels, palate, genitourinary system. The type of malformation produced thus depends on the time of exposure to the teratogen.
The cellular mechanisms by which teratogenic substances produce their effects are not at all well understood. There is a considerable overlap between mutagenicity and teratogenicity. In one large survey, among 78 compounds, 34 were both teratogenic and mutagenic, 19 were negative in both tests and 25 (among them thalidomide) were positive in one but not the other. Damage to DNA is important but, as with carcinogenesis, is not the only factor. The control of morphogenesis is poorly understood; vitamin A derivatives (retinoids) are involved and are potent teratogens (see below). Known teratogens also include several drugs (e.g. methotrexate and phenytoin) that do not react directly with DNA but which inhibit its synthesis by their effects on folate metabolism (see Ch. 25). Administration of folate during pregnancy reduces the frequency of both spontaneous and drug-induced malformations, especially neural tube defects.
The fetus depends on an adequate supply of nutrients during the final stage of histogenesis and functional maturation, and development is regulated by a variety of hormones. Gross structural malformations do not arise from exposure to mutagens at this stage, but drugs that interfere with the supply of nutrients or with the hormonal milieu may have deleterious effects on growth and development. Exposure of a female fetus to androgens at this stage can cause masculinisation. Stilbestrol was commonly given to pregnant women with a history of recurrent miscarriage during the 1950s (for unsound reasons) and causes dysplasia of the vagina of the infant and an increased incidence of carcinoma of the vagina in the teens and twenties. Angiotensin II plays an important part in the later stages of fetal development and in renal function in the fetus, and ACE inhibitors and angiotensin receptor antagonists (’sartans’) cause oligohydramnios and renal failure if administered during later stages of pregnancy and fetal malformations if given earlier.
Testing for Teratogenicity
The thalidomide disaster dramatically brought home the need for routine teratogenicity studies on new therapeutic drugs. Assessment of teratogenicity in humans is a particularly difficult problem for various reasons. One is that the ‘spontaneous’ malformation rate is high (3–10% depending on the definition of a significant malformation) and highly variable between different regions, age groups and social classes. Large-scale studies are required, which take many years and much money to perform, and they usually give suggestive, rather than conclusive, results.
Studies using embryonic stem cells in assessing developmental toxicity are showing some promise (see Bremer & Hartung, 2004, for a review from a regulatory perspective). In vitro methods, based on the culture of cells, organs or whole embryos, have, however, not so far been developed to a level where they satisfactorily predict teratogenesis in vivo, and most regulatory authorities require teratogenicity testing in a rodent plus in one non-rodent species (e.g. rabbit). The visceral yolk sac and development of the chorioallantoic placenta of the rabbit resemble those of humans more so than do those of rodents, in some respects (Foote & Carney, 2000). Pregnant females are dosed at various levels during the critical period of organogenesis, and the fetuses are examined for structural abnormalities. However, poor cross-species correlation means that tests of this kind are not reliably predictive in humans, and it is usually recommended that new drugs are not used in pregnancy unless it is essential.
Some Definite and Probable Human Teratogens
Although many drugs have been found to be teratogenic in varying degrees in experimental animals, relatively few are known to be teratogenic in humans (see Table 57.2). Some of the more important are discussed below.
Thalidomide
Thalidomide is virtually unique in producing, at therapeutic dosage, virtually 100% malformed infants when taken in the first 3–6 weeks of gestation. It was introduced in 1957 as a hypnotic and sedative with the special feature that it was much less hazardous in overdosage than barbiturates, and it was even recommended specifically for use in pregnancy (with the advertising slogan ‘the safe hypnotic’). It had been subjected to toxicity testing only in mice, which are resistant to thalidomide teratogenicity (probably because mouse embryonic cells have higher glutathione levels than humans; Knobloch et al., 2008). Thalidomide was marketed energetically and successfully, and the first suspicion of its teratogenicity arose early in 1961 with reports of a sudden increase in the incidence of phocomelia. This abnormality (‘seal limbs’) consists of an absence of development of the long bones of the arms and legs, and had hitherto been virtually unknown. At this time, approximately 1 000 000 tablets were being sold daily in West Germany. Reports of phocomelia came simultaneously from Hamburg and Sydney, and the connection with thalidomide was made.6 The drug was withdrawn late in 1961, by which time an estimated 10 000 malformed babies had been born (Fig. 57.3 illustrates the use of data linkage in detecting delayed ADRs). Despite intensive study, its mechanism remains poorly understood, although epidemiological investigation showed very clearly the correlation between the time of exposure and the type of malfunction produced (Table 57.4).
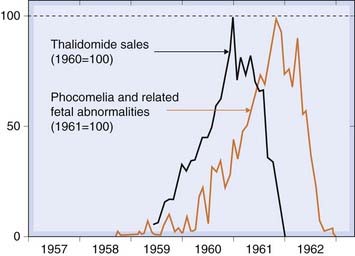
Fig. 57.3 Incidence of major fetal abnormalities in Western Europe following the introduction and withdrawal of thalidomide linked to sales data for thalidomide.
Table 57.4 Thalidomide teratogenesis
| Day of gestation | Type of deformity |
|---|---|
| 21–22 | Malformation of ears Cranial nerve defects |
| 24–27 | Phocomelia of arms |
| 28–29 | Phocomelia of arms and legs |
| 30–36 | Malformation of hands Anorectal stenosis |
Cytotoxic drugs
Many alkylating agents (e.g. chlorambucil and cyclophosphamide) and antimetabolites (e.g. azathioprine and mercaptopurine) cause malformations when used in early pregnancy but more often lead to abortion (see Ch. 55). Folate antagonists (e.g. methotrexate) produce a much higher incidence of major malformations, evident in both live-born and stillborn fetuses.
Retinoids
Etretinate, a retinoid (i.e. vitamin A derivative) with marked effects on epidermal differentiation, is a known teratogen and causes a high proportion of serious abnormalities (notably skeletal deformities) in exposed fetuses. Dermatologists use retinoids to treat skin diseases including several, such as acne and psoriasis, that are common in young women. Etretinate accumulates in subcutaneous fat and is eliminated extremely slowly, detectable amounts persisting for many months after chronic dosing is discontinued. Because of this, women should avoid pregnancy for at least 2 years after treatment. Acitretin is an active metabolite of etretinate. It is equally teratogenic, but tissue accumulation is less pronounced and elimination may be more rapid.
Heavy metals
Lead, cadmium and mercury all cause fetal malformation in humans. The main evidence comes from Minamata disease, named after the locality in Japan where an epidemic occurred when the local population ate fish contaminated with methylmercury that had been used as an agricultural fungicide. This impaired brain development in exposed fetuses, resulting in cerebral palsy and mental retardation, often with microcephaly. Mercury, like other heavy metals, inactivates many enzymes by forming covalent bonds with sulfhydryl and other groups, and this is believed to be responsible for these developmental abnormalities.
Antiepileptic drugs
Congenital malformations are increased two- to three-fold in babies of epileptic mothers. Interestingly, all existing antiepileptic drugs have been implicated, including phenytoin (particularly cleft lip/palate), valproate (neural tube defects) and carbamazepine (spina bifida and hypospadias, a malformation of the male urethra), as well as newer agents including lamotrigine (Ch. 44).
Warfarin
Administration of warfarin (Ch. 24) in the first trimester is associated with nasal hypoplasia and various central nervous system abnormalities, affecting roughly 25% of exposed babies. In the last trimester, it must not be used because of the risk of intracranial haemorrhage in the baby during delivery.
Assessment of Genotoxic Potential
Registration of pharmaceuticals requires a comprehensive assessment of their genotoxic potential. Because no single test is adequate, the usual approach recommended by the International Conference on Harmonisation (ESRA Rapporteur 1997 4: 5–7) is to carry out a battery of in vitro and in vivo tests for genotoxicity. The following battery is often used:
Teratogenesis and drug-induced fetal damage
Allergic Reactions to Drugs
Allergic reactions of various kinds are a common form of adverse response to drugs. Most drugs, being low-molecular-weight substances, are not immunogenic in themselves. A drug or its metabolites can, however, act as a hapten by interacting with protein to form a stable conjugate that is immunogenic (Ch. 6). The immunological basis of some allergic drug reactions has been well worked out, but often it is inferred from the clinical characteristics of the reaction, and direct evidence of an immunological mechanism is lacking. Suggestive features are as follow:
The overall incidence of allergic drug reactions is variously reported as being between 2% and 25%. Most are minor skin eruptions. Serious reactions (e.g. anaphylaxis, haemolysis and bone marrow depression) are rare. Penicillins, which are the commonest cause of drug-induced anaphylaxis, produce this response in an estimated 1 in 50 000 patients exposed. Rashes can be severe, and fatalities occur with Stevens–Johnson syndrome (provoked, for example, by sulfonamides) and toxic epidermal necrolysis (TEN, which can be caused by allopurinol). The association between cabamazepine-induced TEN and the gene for a particular HLA allele HLAB*1502 in people of Asian ancestry is mentioned in Chapter 11. Susceptibility to severe rashes in response to abacavir is closely linked to the human leukocyte antigen (HLA) variant HLAB*5701 and this forms the basis of a clinically useful genomic test (Ch. 11).
Immunological Mechanisms
The formation of an immunogenic conjugate between a small molecule and an endogenous protein requires covalent bonding. In most cases, reactive metabolites, rather than the drug itself, are responsible. Such reactive metabolites can be produced during drug oxidation or by photoactivation in the skin. They may also be produced by the action of toxic oxygen metabolites generated by activated leukocytes. Rarely (e.g. in drug-induced lupus erythematosus), the reactive moiety interacts to form an immunogen with nuclear components (DNA, histone) rather than proteins (see below). Conjugation with a macromolecule is usually essential, although penicillin is an exception because it can form sufficiently large polymers in solution to elicit an anaphylactic reaction in a sensitised individual even without conjugation to protein, although penicillin–protein conjugates can also act as the immunogen.
Clinical Types of Allergic Response to Drugs
In the Gell and Coombs classification of hypersensitivity reactions (Ch. 6), types I, II and III are antibody-mediated reactions and type IV is cell mediated. Unwanted reactions to drugs involve both antibody- and cell-mediated reactions. The more important clinical manifestations of hypersensitivity include anaphylactic shock, haematological reactions, allergic liver damage and other hypersensitivity reactions.
Anaphylactic Shock
Anaphylactic shock—see also Chapter 27—is a type I hypersensitivity response. It is a sudden and life-threatening reaction that results from the release of histamine, leukotrienes and other mediators. The main features include urticarial rash, swelling of soft tissues, bronchoconstriction and hypotension.
Penicillins account for about 75% of anaphylactic deaths, reflecting the frequency with which they are used in clinical practice. Other drugs that can cause anaphylaxis include various enzymes, for example asparaginase (Ch. 55); therapeutic monoclonal antibodies (Ch. 59), hormones, for example corticotropin (adrenocorticotrophic hormone; Ch. 32); heparin (Ch. 24); dextrans; radiological contrast agents; vaccines; and other serological products. Anaphylaxis with local anaesthetics (Ch. 42), the antiseptic chlorhexidine and with many other drugs (sometimes as a consequence of contaminants such as latex used to seal reusable vials or of excipients and colouring agents rather than the drug itself) can occur. Treatment of anaphylaxis is given in Chapter 27.
It is sometimes feasible to carry out a skin test for the presence of anaphylactic hypersensitivity, which involves injecting a minute dose intradermally. A patient who reports that she or he is allergic to a drug such as penicillin may actually be allergic to fungal contaminants in early preparations rather than to penicillin itself. The use of penicilloylpolylysine as a skin test reagent for penicillin allergy is an improvement over the use of penicillin itself, because it bypasses the need for conjugation of the test substance, thereby reducing the likelihood of a false negative. Other specialised tests are available to detect the presence of specific immunoglobulin E in the plasma, or to measure histamine release from the patient’s basophils, but these are not used routinely.
Other drug-induced type I hypersensitivity reactions include bronchospasm (Ch. 27) and urticaria.
Haematological Reactions
Drug-induced haematological reactions can be produced by type II, III or IV hypersensitivity. Type II reactions can affect any or all of the formed elements of the blood, which may be destroyed by effects either on the circulating blood cells themselves or on their progenitors in the bone marrow. They involve antibody binding to a drug–macromolecule complex on the cell surface membrane. The antigen–antibody reaction activates complement, leading to lysis, or provokes attack by killer lymphocytes or phagocytic leukocytes (Ch. 6). Haemolytic anaemia has been most commonly reported with sulfonamides and related drugs (Ch. 50) and with an antihypertensive drug, methyldopa (Ch. 14), which is still widely used to treat hypertension during pregnancy. With methyldopa, significant haemolysis occurs in less than 1% of patients, but the appearance of antibodies directed against the surface of red cells is detectable in 15% by the Coombs test. The antibodies are directed against Rh antigens, but it is not known how methyldopa produces this effect.
Drug-induced agranulocytosis (complete absence of circulating neutrophils) is usually delayed 2–12 weeks after beginning drug treatment but may then be sudden in onset. It often presents with mouth ulcers, a severe sore throat or other infection. Serum from the patient lyses leukocytes from other individuals, and circulating antileukocyte antibodies can usually be detected immunologically. Drugs associated with agranulocytosis include NSAIDs, especially phenylbutazone (Ch. 26), carbimazole (Ch. 33) and clozapine (Ch. 45) (increased genetic susceptibility associated with HLA-DQB1*0201 is mentioned in Ch. 11) and sulfonamides and related drugs (e.g. thiazides and sulfonylureas). Agranulocytosis is rare but life-threatening. Recovery when the offending drug is stopped is often slow or absent. Antibody-mediated leukocyte destruction must be distinguished from the direct effect of cytotoxic drugs (see Ch. 55), which cause granulocytopenia that is rapid in onset, predictably related to dose and reversible.
Thrombocytopenia (reduction in platelet numbers) can be caused by type II reactions to quinine (Ch. 53), heparin (Ch. 24) and thiazide diuretics (Ch. 28).
Some drugs (notably chloramphenicol) can suppress all three haemopoietic cell lineages, giving rise to aplastic anaemia (anaemia with associated agranulocytosis and thrombocytopenia).
The distinction between type III and type IV hypersensitivity reactions in the causation of haematological reactions is not clear-cut, and either or both mechanisms can be involved.
Allergic Liver Damage
Most drug-induced liver damage results from the direct toxic effects of drugs or their metabolites, as described above. However, hypersensitivity reactions are sometimes involved, a particular example being halothane-induced hepatic necrosis (see Ch. 40). Trifluoracetylchloride, a reactive metabolite of halothane, couples to a macromolecule to form an immunogen. Most patients with halothane-induced liver damage have antibodies that react with halothane–carrier conjugates. Halothane–protein antigens can be expressed on the surface of hepatocytes. Destruction of the cells occurs by type II hypersensitivity reactions involving killer T cells, and type III reactions can also contribute.
Other Hypersensitivity Reactions
The clinical manifestations of type IV hypersensitivity reactions are diverse, ranging from minor skin rashes to generalised autoimmune disease. Fever may accompany these reactions. Rashes can be antibody mediated but are usually cell mediated. They range from mild eruptions to fatal exfoliation. Stevens–Johnson syndrome is a very severe generalised rash that extends into the alimentary tract and carries an appreciable mortality. In some cases, the lesions are photosensitive, probably because ultraviolet light converts the drug to reactive products.
Some drugs (notably hydralazine and procainamide) can produce an autoimmune syndrome resembling systemic lupus erythematosus. This is a multisystem disorder in which there is immunological damage to many organs and tissues (including joints, skin, lung, central nervous system and kidney) caused particularly, but not exclusively, by type III hypersensitivity reactions. The prodigious array of antibodies directed against ‘self’ components has been termed an ‘autoimmune thunderstorm’. The antibodies react with determinants shared by many molecules, for example the phosphodiester backbone of DNA, RNA and phospholipids. In drug-induced systemic lupus erythematosus, the immunogen may result from the reactive drug moiety interacting with nuclear material, and joint and pulmonary damage is common. The condition usually resolves when treatment with the offending drug is stopped.
Allergic reactions to drugs
References and Further Reading
Aronson J.K., Ferner R.E. Joining the DoTS: a new approach to classifying adverse drug reactions. Br. Med. J.. 2003;327:1222-1225. (Description of ADRs in terms of dose, time course and susceptibility)
Pirmohamed M., James S., Meakin S., et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. Br. Med. J.. 2004;329:15-19. (There were 1225 admissions related to an adverse drug reaction. The median bed stay was 8 days, accounting for 4% of the hospital bed capacity. The projected annual cost is £466 million. Most reactions were avoidable. Drugs most commonly implicated were aspirin and other NSAIDs, diuretics, warfarin; the most common reaction was gastrointestinal bleeding)
Rawlins M.D., Thomson J.W. Mechanisms of adverse drug reactions. In: Davies D.M., editor. Textbook of adverse drug reactions. third ed. Oxford: Oxford University Press; 1985:12-38. (Type A/type B classification)
Drug toxicity: general and mechanistic aspects
Bhogal N., Grindon C., Combes R., Balls M. Toxicity testing: creating a revolution based on new technologies. Trends Biotechnol.. 2005;23:299-307. (Reviews current and likely future value of new technologies in relation to toxicological evaluation)
Bremer S., Hartung T. The use of embryonic stem cells for regulatory developmental toxicity testing in vitro—the current status of test development. Curr. Pharm. Des.. 2004;10:2733-2747. (Summarises requirements for an in vitro embryotoxicity test needed for regulatory toxicity testing)
Parng C. In vivo zebrafish assays for toxicity testing. Curr. Opin. Drug Discov. Devel.. 2005;8:100-106. (Effective in vivo toxicity screening early in development can reduce the number of compounds that progress to laborious and costly late-stage animal testing. The transparent zebra fish provides accessibility to internal organs, tissues and even cells, and has emerged as a model organism for toxicity testing. Straightforward in vivo zebra fish assays can serve as an intermediate step between cell-based and mammalian testing, thus streamlining the drug development timeline)
Pirmohamed M. Drug-induced apoptosis: clinical significance. Drug Metab. Rev.. 35, 2003. 24-24. 48 (Suppl. 1)
Pirmohamed M. Role of the immune system in idiosyncratic drug reactions. Drug Metab. Rev.. 36, 2004. 29-29. 58 (Suppl. 1)
Timbrell J.A. Principles of biochemical toxicity. New York: Informa Healthcare; 2009.
Uetrecht J. Screening for the potential of a drug candidate to cause idiosyncratic drug reactions. Drug Discov. Today. 2003;8:832-837. (Highlights current mechanistic hypotheses of idiosyncratic drug reactions and discusses future directions in the development of better predictive tests)
Walker D.K. The use of pharmacokinetic and pharmacodynamic data in the assessment of drug safety in early drug development. Br. J. Clin. Pharm.. 2004;58:601-608. (Pharmacokinetic profile is a factor in assessing safety during early drug development, especially in relation to safety parameters such as QT interval prolongation, where free plasma concentrations are predictive; procedures are available that allow this on the microdose scale—potential limitations are discussed)
Drug toxicity: carcinogenesis, teratogenesis
Briggs G.G., Freeman R.K., Yaffe S.J. Drugs in pregnancy and lactation, eighth ed. Philadelphia: Lippincott, Williams & Wilkins; 2008. (Invaluable reference guide to fetal and neonatal risk for clinicians caring for pregnant women)
Collins M.D., Mayo G.E. Teratology of retinoids. Annu. Rev. Pharmacol. Toxicol.. 1999;39:399-430. (Overviews principles of teratology as they apply to the retinoids, describes signal transduction of retinoids and toxikinetics)
Foote R.H., Carney E.W. The rabbit as a model for reproductive and developmental toxicity studies. Reprod. Toxicol.. 2000;14:477-493. (Discusses the use of the rabbit in developmental toxicity and teratology studies)
Knobloch J., Reimann K., Klotz L.-O., Rüther U. Thalidomide resistance is based on the capacity of the glutathione-dependent antioxidant defense. Mol. Pharm.. 2008;5:1138-1144.
Sjöström H., Nilsson R. Thalidomide and the power of the drug companies. London: Penguin Books; 1972.
Drug toxicity: organ involvement
Murray M.C., Brater D.C. Renal toxicity of the nonsteroidal anti-inflammatory drugs. Annu. Rev. Pharmacol. Toxicol.. 1993;33:435-465.
Park B.K., Kitteringham N.R., Maggs J.L., et al. The role of metabolic activation in drug-induced hepatotoxicity. Annu. Rev. Pharmacol. Toxicol.. 2005;45:177-202. (Reviews evidence for reactive metabolite formation from hepatotoxic drugs such as paracetamol, tamoxifen, diclofenac and troglitazone, and the current hypotheses of how this leads to liver injury)
Ritter J.M., Harding I., Warren J.B. Precaution, cyclooxygenase inhibition, and cardiovascular risk. Trends Pharmacol. Sci.. 2009;30:503-514.
Svensson C.K., Cowen E.W., Gaspari A.A. Cutaneous drug reactions. Pharmacol. Rev.. 2001;53:357-380. (Covers epidemiology, clinical morphology and mechanisms. Assesses current knowledge of four types of cutaneous drug reaction: immediate-type immune mediated, delayed-type immune mediated, photosensitivity and autoimmune. Also reviews the role of viral infection as predisposing factor)
Valentin J.-P. Reducing QT liability and proarrhythmic risk in drug discovery and development. Br. J. Pharmacol.. 2010;159:5-11. (See also accompanying articles in this themed section on QT safety)
16.5% of hospital admissions were due to ADRs at a projected annual cost of £466 million in the UK. Antiplatelet drugs, diuretics, non-steroidal anti-inflammatory drugs and anticoagulants between them accounted for 50% of the ADRs. Most events were avoidable and 2.3% of the patients died.
2Hesitation in prescribing a newly licensed drug may delay recognition of an ADR without reducing the total number of patients harmed, so a ‘cautious physician’ is one who prefers to let others take the risk. Grant of a product licence to a company to market a new drug does not require evidence of superiority over existing treatments, so from the patient’s perspective a physician who is neither at the forefront of fashion nor the last to adopt a genuine advance may be the best bet in an uncertain world.
3The value of toxicity testing is illustrated by experience with triparanol, a cholesterol-lowering drug marketed in the USA in 1959. Three years later, a team from the Food and Drug Administration, acting on a tip-off, paid the manufacturer a surprise visit that revealed falsification of toxicology data demonstrating cataracts in rats and dogs. The drug was withdrawn, but some patients who had been taking it for a year or more also developed cataracts. Regulatory authorities now require that toxicity testing is performed under a tightly defined code of practice (Good Laboratory Practice), which incorporates many safeguards to minimise the risk of error or fraud.
4Aficionados of Dennis Potter will recall the protagonist in the television drama The Singing Detective; Potter was himself afflicted by the most severe form of the disease.
5The renal papilla is the part of the kidney exposed to the highest concentration of solutes, including drug metabolites; it also has a lower effective blood flow than other parts as a result of counter-current exchange in the vasa recta.
6A severe peripheral neuropathy, leading to irreversible paralysis and sensory loss, was reported within a year of the drug’s introduction and subsequently confirmed in many reports. The drug company responsible was less than punctilious in acting on these reports (see Sjöström & Nilsson, 1972), which were soon eclipsed by the discovery of teratogenic effects, but the neurotoxic effect was severe enough in its own right to have necessitated withdrawal of the drug from general use. Today, use of thalidomide has had a resurgence related to several highly specialised applications. It is prescribed by specialists (in dermatology, oncology and in HIV infection, among others) under tightly controlled and restricted conditions.