30 The control of blood glucose and drug treatment of diabetes mellitus
Overview
In this chapter, we describe the endocrine control of blood glucose by pancreatic hormones, especially insulin but also glucagon, somatostatin and amylin, and gut hormones (incretins), especially glucagon-like peptide-1 (GLP-1) and gastric inhibitory peptide (GIP, which is also known as glucose-dependent insulinotropic peptide). The second part of the chapter is devoted to diabetes mellitus and its treatment with insulin preparations (including insulin analogues), and other hypoglycaemic agents—metformin, sulfonylureas, α-glucosidase inhibitors, glitazones, long-acting incretin mimetics such as exenatide, and gliptins which potentiate incretins by blocking their degradation.
Introduction
Insulin is the main hormone controlling intermediary metabolism. Its most striking acute effect is to lower blood glucose. Reduced (or absent) secretion of insulin often coupled with reduced sensitivity to its action, ‘insulin resistance’ which is closely related to obesity, causes diabetes mellitus. This disease, recognised since ancient times, is named for the production of copious volumes of sugary urine. Diabetes is rapidly increasing to epidemic proportions (in step with obesity, Ch. 31), and its consequences are dire—especially atherosclerosis (myocardial and cerebral infarction, amputation), kidney failure, neuropathy and blindness.
In this chapter, we first describe the control of blood sugar. The second part of the chapter is devoted to diabetes mellitus and its treatment with drugs.
Control of Blood Glucose
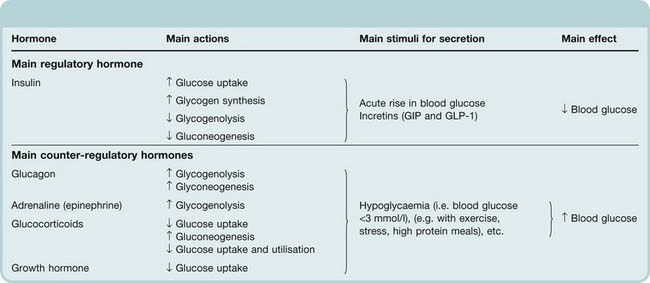
Glucose is the obligatory source of energy for the adult brain, and physiological control of blood glucose reflects the need to maintain adequate fuel supplies in the face of intermittent food intake and variable metabolic demands. More fuel is made available by feeding than is required immediately, and excess calories are stored as glycogen or fat. During fasting, these energy stores need to be mobilised in a regulated manner. The most important regulatory hormone is insulin, the actions of which are described below. Increased blood glucose stimulates insulin secretion (Fig. 30.1), whereas reduced blood glucose reduces insulin secretion. The effect of glucose on insulin secretion depends on whether the glucose load is administered intravenously or by mouth. Glucose administered by mouth is more effective in stimulating insulin secretion because it stimulates the release of incretin hormones from the gut, which promote insulin secretion (Fig. 30.1). The effect of glucose on insulin secretion is abnormal in patients with diabetes (Fig. 30.2). Hypoglycaemia, caused by excessive insulin, not only reduces insulin secretion but also elicits secretion of an array of ‘counter-regulatory’ hormones, including glucagon, adrenaline (Ch. 14), glucocorticoids (Ch. 32) and growth hormone (Ch. 32), all of which increase blood glucose. Their main effects on glucose uptake and carbohydrate metabolism are summarised and contrasted with those of insulin in Table 30.1.
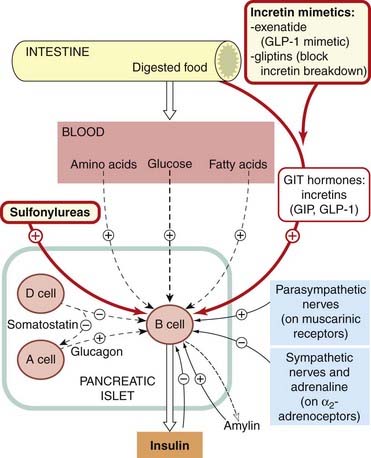
Fig. 30.1 Factors regulating insulin secretion.
Blood glucose is the most important factor. Drugs used to stimulate insulin secretion are shown in red-bordered boxes. Glucagon potentiates insulin release but opposes some of its peripheral actions and increases blood glucose. GIP, gastric inhibitory peptide; GIT, gastrointestinal tract; GLP-1, glucagon-like peptide-1.
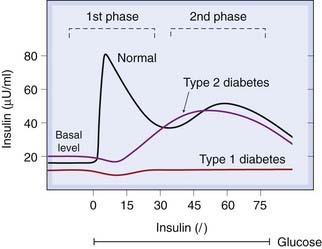
Fig. 30.2 Schematic diagram of the two-phase release of insulin in response to a constant glucose infusion.
The first phase is missing in type 2 (non-insulin-dependent) diabetes mellitus, and both are missing in type 1 (insulin-dependent) diabetes mellitus. The first phase is also produced by amino acids, sulfonylureas, glucagon and gastrointestinal tract hormones.
(Data from Pfeifer et al. 1981 Am J Med 70: 579–588.)
Table 30.1 The effect of hormones on blood glucose
Pancreatic Islet Hormones
The islets of Langerhans, the endocrine part of the pancreas, contain four main types of peptide-secreting cells: B (or β) cells secrete insulin, A cells secrete glucagon, D cells secrete somatostatin and PP cells secrete pancreatic polypeptide (the function of which is unknown). The core of each islet contains mainly the predominant B cells surrounded by a mantle of A cells interspersed with D cells or PP cells (see Fig. 30.1). In addition to insulin, B cells secrete a peptide known as islet amyloid polypeptide or amylin, which delays gastric emptying and opposes insulin by stimulating glycogen breakdown in striated muscle, and C-peptide (see below). Glucagon opposes insulin, increasing blood glucose and stimulating protein breakdown in muscle. Somatostatin inhibits secretion of insulin and of glucagon. It is widely distributed outside the pancreas and is also released from the hypothalamus, inhibiting the release of growth hormone from the pituitary gland (Ch. 32).
Insulin
Insulin was the first protein for which the amino acid sequence was determined (by Sanger’s group in Cambridge in 1955). It consists of two peptide chains (of 21 and 30 amino acid residues) linked by disulfide bonds.
Synthesis and Secretion
Like other peptide hormones (see Ch. 19), insulin is synthesised as a precursor (preproinsulin) in the rough endoplasmic reticulum. Preproinsulin is transported to the Golgi apparatus, where it undergoes proteolytic cleavage to proinsulin and then to insulin plus a fragment of uncertain function called C-peptide.1 Insulin and C-peptide are stored in granules in B cells, and are normally co-secreted by exocytosis in equimolar amounts together with smaller and variable amounts of proinsulin.
The main factor controlling the synthesis and secretion of insulin is the blood glucose concentration (Fig. 30.1). B cells respond both to the absolute glucose concentration and to the rate of change of blood glucose. Other physiological stimuli to insulin release include amino acids (particularly arginine and leucine), fatty acids, the parasympathetic nervous system and incretins (see below). The main incretins are GLP-1 and GIP. Pharmacologically, sulfonylurea drugs (see below) act by releasing insulin.
There is a steady basal release of insulin and also a response to an increase in blood glucose. This response has two phases: an initial rapid phase reflecting release of stored hormone, and a slower, delayed phase reflecting continued release of stored hormone and new synthesis (Fig. 30.2). The response is abnormal in diabetes mellitus, as discussed later.
ATP-sensitive potassium channels (KATP; Ch. 4) determine the resting membrane potential in B cells. Glucose enters B cells via a membrane transporter called Glut-2, and its subsequent metabolism via glucokinase (the rate-limiting enzyme that acts as the ‘glucose sensor’ linking insulin secretion to extracellular glucose) and glycolysis increases intracellular ATP. This blocks KATP channels, causing membrane depolarisation and opening of voltage-dependent calcium channels, leading to Ca2+ influx. The resulting increase in cytoplasmic Ca2+ triggers insulin secretion, but only in the presence of amplifying messengers including diacylglycerol, non-esterified arachidonic acid (which facilitates further Ca2+ entry), and 12-lipoxygenase products of arachidonic acid (mainly 12-S-hydroxyeicosatetraenoic acid or 12-S-HETE; see Ch. 17). Phospholipases are commonly activated by Ca2+, but free arachidonic acid is liberated in B cells by an ATP-sensitive Ca2+-insensitive (ASCI) phospholipase A2. Consequently, in B cells, Ca2+ entry and arachidonic acid production are both driven by ATP, linking cellular energy status to insulin secretion.
Insulin release is inhibited by the sympathetic nervous system (Fig. 30.1). Adrenaline (epinephrine) increases blood glucose by inhibiting insulin release (via α2 adrenoceptors) and by promoting glycogenolysis via β2-adrenoceptors in striated muscle and liver. Several peptides, including somatostatin, galanin (an endogenous KATP activator) and amylin, also inhibit insulin release.
About one-fifth of the insulin stored in the pancreas of the human adult is secreted daily. Circulating insulin is measured by immunoassay, but this may give an overestimate because many insulin antibodies cross-react with proinsulin and its less active degradation products. The plasma insulin concentration after an overnight fast is 20–50 pmol/l. Plasma insulin concentration is reduced in patients with type 1 (insulin-dependent) diabetes mellitus (see below), and markedly increased in patients with insulinomas (uncommon functioning tumours of B cells), as is C-peptide, with which it is co-released.2 It is also raised in obesity and other normoglycaemic insulin-resistant states.
Actions
Insulin is the main hormone controlling intermediary metabolism, having actions on liver, fat and muscle (Table 30.2). It is an anabolic hormone: its overall effect is to conserve fuel by facilitating the uptake and storage of glucose, amino acids and fats after a meal. Acutely, it reduces blood glucose. Consequently, a fall in plasma insulin increases blood glucose. The biochemical pathways through which insulin exerts its effects are summarised in Figure 30.3, and molecular aspects of its mechanism are discussed below.
Table 30.2 Effects of insulin on carbohydrate, fat and protein metabolism
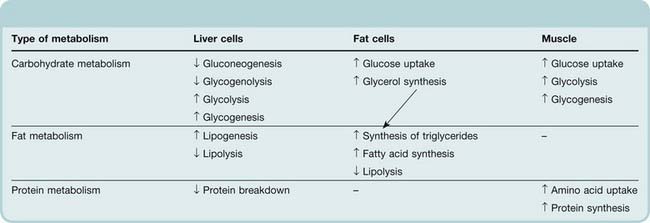
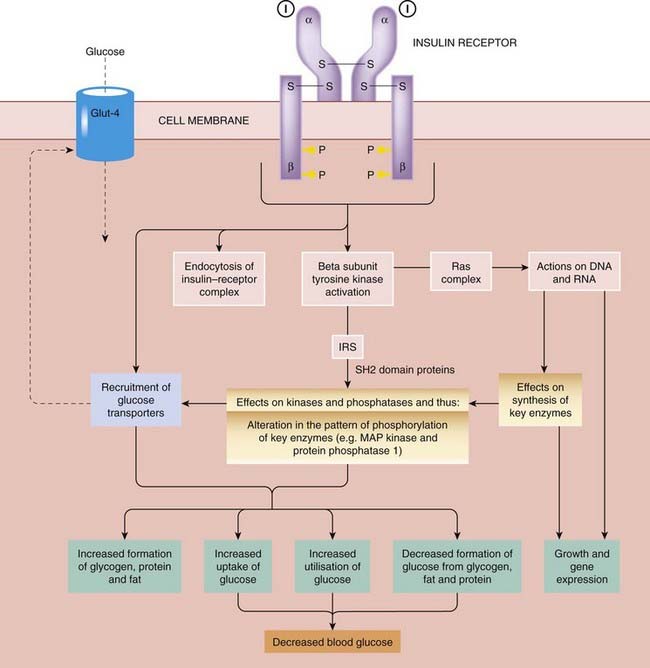
Fig. 30.3 Insulin signalling pathways.
I, insulin; Glut-4, an insulin-sensitive glucose transporter present in muscle and fat cells; IRS, insulin receptor substrate (several forms: 1–4).
Insulin influences glucose metabolism in most tissues, especially the liver, where it inhibits glycogenolysis (glycogen breakdown) and gluconeogenesis (synthesis of glucose from non-carbohydrate sources) while stimulating glycogen synthesis. It also increases glucose utilisation (glycolysis), but the overall effect is to increase hepatic glycogen stores.
In muscle, unlike liver, uptake of glucose is slow and is the rate-limiting step in carbohydrate metabolism. The main effects of insulin are to increase facilitated transport of glucose via a transporter called Glut-4, and to stimulate glycogen synthesis and glycolysis.
Insulin increases glucose uptake by Glut-4 in adipose tissue as well as in muscle, enhancing glucose metabolism. One of the main end products of glucose metabolism in adipose tissue is glycerol, which is esterified with fatty acids to form triglycerides, thereby affecting fat metabolism (see below and Table 30.2).
Insulin increases synthesis of fatty acid and triglyceride in adipose tissue and in liver. It inhibits lipolysis, partly via dephosphorylation—and hence inactivation—of lipases (Table 30.2). It also inhibits the lipolytic actions of adrenaline, growth hormone and glucagon by opposing their actions on adenylyl cyclase.
Insulin stimulates uptake of amino acids into muscle and increases protein synthesis. It also decreases protein catabolism and inhibits oxidation of amino acids in the liver.
Other metabolic effects of insulin include transport into cells of K+, Ca2+, nucleosides and inorganic phosphate.3
Long-term effects of insulin
In addition to its rapid effects on metabolism, exerted via altered activity of enzymes and transport proteins, insulin has long-term actions via altered enzyme synthesis. It is an important anabolic hormone during fetal development. It stimulates cell proliferation and is implicated in somatic and visceral growth and development.
Mitogenic actions of insulin are of great concern in the development of insulin analogues, because these are intended for long-term use; insulin glargine (one widely used analogue; see below) is 6–8-fold more mitogenic than human insulin, and cultured breast cancer cells proliferate in response to near-therapeutic concentrations of this analogue in vitro, but it is not known if there is any clinically significant parallel in vivo. Mammary tumours developed in rats given one long-acting insulin analogue.
Mechanism of Action
Insulin binds to a specific receptor on the surface of its target cells. The receptor is a large transmembrane glycoprotein complex belonging to the tyrosine kinase-linked type 3 receptor superfamily (Ch. 3) and consisting of two α and two β subunits (Fig. 30.3). Occupied receptors aggregate into clusters, which are subsequently internalised in vesicles, resulting in downregulation. Internalised insulin is degraded in lysosomes, but the receptors are recycled to the plasma membrane.
 The signal transduction mechanisms that link receptor binding to the biological effects of insulin are complex. Receptor autophosphorylation—the first step in signal transduction—is a consequence of dimerisation, allowing each receptor to phosphorylate the other, as explained in Chapter 3.
The signal transduction mechanisms that link receptor binding to the biological effects of insulin are complex. Receptor autophosphorylation—the first step in signal transduction—is a consequence of dimerisation, allowing each receptor to phosphorylate the other, as explained in Chapter 3.
Insulin receptor substrate (IRS) proteins undergo rapid tyrosine phosphorylation specifically in response to insulin and insulin-like growth factor-1 but not to other growth factors. The best-characterised substrate is IRS-1, which contains 22 tyrosine residues that are potential phosphorylation sites. It interacts with proteins that contain a so-called SH2 domain (see Ch. 3, Fig. 3.15), thereby passing on the insulin signal. Knockout mice lacking IRS-1 are hyporesponsive to insulin (insulin resistant) but do not become diabetic, because of robust B-cell compensation with increased insulin secretion. By contrast, mice lacking IRS-2 fail to compensate and develop overt diabetes, implicating the IRS-2 gene as a candidate for human type 2 diabetes (IRS proteins are reviewed by Lee & White, 2004). Activation of phosphatidylinositol 3-kinase by interaction of its SH2 domain with phosphorylated IRS has several important effects, including recruitment of insulin-sensitive glucose transporters (Glut-4) from the Golgi apparatus to the plasma membrane in muscle and fat cells.
The longer-term actions of insulin entail effects on DNA and RNA, mediated partly at least by the Ras signalling complex. Ras is a protein that regulates cell growth and cycles between an active GTP-bound form and an inactive GDP-bound form (see Chs 3 and 55). Insulin shifts the equilibrium in favour of the active form, and initiates a phosphorylation cascade that results in activation of mitogen-activated protein kinase (MAP-kinase), which in turn activates several nuclear transcription factors, leading to the expression of genes that are involved both with cell growth and with intermediary metabolism. Regulation of the rate of mRNA transcription by insulin provides an important means of modulating enzyme activity.
Insulin for treatment of diabetes mellitus is considered below.
Glucagon
Synthesis and Secretion
Glucagon is a single-chain polypeptide of 21 amino acid residues synthesised mainly in the A cell of the islets, but also in the upper gastrointestinal tract. It has considerable structural homology with other gastrointestinal tract hormones, including secretin, vasoactive intestinal peptide and GIP (see Ch. 29).
One of the main physiological stimuli to glucagon secretion is the concentration of amino acids, in particular L-arginine, in plasma. Therefore an increase in secretion follows ingestion of a high-protein meal, but compared with insulin there is relatively little change in plasma glucagon concentrations throughout the day. Glucagon secretion is stimulated by low and inhibited by high concentrations of glucose and fatty acids in the plasma. Sympathetic nerve activity and circulating adrenaline stimulate glucagon release via β-adrenoceptors. Parasympathetic nerve activity also increases secretion, whereas somatostatin, released from D cells adjacent to the glucagon-secreting A cells in the periphery of the islets, inhibits glucagon release.
Endocrine pancreas and blood glucose 
Actions
Glucagon increases blood glucose and causes breakdown of fat and protein. It acts on specific G-protein-coupled receptors to stimulate adenylyl cyclase, and consequently its actions are somewhat similar to β-adrenoceptor-mediated actions of adrenaline. Unlike adrenaline, however, its metabolic effects are more pronounced than its cardiovascular actions. Glucagon is proportionately more active on liver, while the metabolic actions of adrenaline are more pronounced on muscle and fat. Glucagon stimulates glycogen breakdown and gluconeogenesis, and inhibits glycogen synthesis and glucose oxidation. Its metabolic actions on target tissues are thus the opposite of those of insulin. Glucagon increases the rate and force of contraction of the heart, although less markedly than adrenaline.
Clinical uses of glucagon are summarised in the clinical box.
Somatostatin
Somatostatin is secreted by the D cells of the islets. It is also generated in the hypothalamus, where it acts to inhibit the release of growth hormone (see Ch. 32). In the islet, it inhibits release of insulin and of glucagon. Octreotide is a long-acting analogue of somatostatin. It inhibits release of a number of hormones, and is used clinically to relieve symptoms from several uncommon gastroenteropancreatic endocrine tumours, and for treatment of acromegaly4 (the endocrine disorder caused by a functioning tumour of cells that secrete growth hormone from the anterior pituitary; see Ch. 32).
Amylin (Islet Amyloid Polypeptide)
The term amyloid refers to amorphous protein deposits in different tissues that occur in a variety of diseases, including several neurodegenerative conditions (see Ch. 39). Amyloid deposits occur in the pancreas of patients with diabetes mellitus, although it is not known if this is functionally important. The major component of pancreatic amyloid is a 37-amino acid residue peptide known as islet amyloid polypeptide or amylin. This is stored with insulin in secretory granules in B cells and is co-secreted with insulin. Amylin delays gastric emptying. Supraphysiological concentrations stimulate the breakdown of glycogen to lactate in striated muscle. Amylin also inhibits insulin secretion (Fig. 30.1). It is structurally related to calcitonin (see Ch. 35) and has weak calcitonin-like actions on calcium metabolism and osteoclast activity. It is also about 50% identical with calcitonin gene-related peptide (CGRP; see Ch. 19), and large intravenous doses cause vasodilatation, presumably by an action on CGRP receptors. Whether amylin has a role in the physiological control of glucose metabolism is controversial, but there is interest in the therapeutic potential of amylin agonists (such as pramlintide, an analogue with three proline substitutions that reduce its tendency to aggregate into insoluble fibrils)—see Schmitz et al. (2004) for a review.
Incretins
La Barre suggested in the 1930s that crude secretin contained two active principles: ‘excretin’, which stimulates the exocrine pancreas and ‘incretin’, which stimulates insulin release. He proposed that incretin presented possibilities for the treatment of diabetes. ‘Excretin’ did not catch on (perhaps not helped by an unfortunate association with other bodily functions—at least to an Anglo-Saxon ear), but ‘incretin’ has gone from strength to strength, and some 80 years later several incretin-based drugs are now licensed for clinical use (see below). Incretin action proved to be due to peptide hormones released from the gut, mainly glucagon-like insulinotropic peptide (GIP) and glucagon-like peptide-1 (GLP-1). These are both members of the glucagon peptide superfamily (Ch. 19). GIP is a 42-amino acid peptide stored in and secreted by enteroendocrine K cells in the duodenum and proximal jejunum. GLP-1 is secreted by L cells which are more widely distributed in the gut, including in the ileum and colon as well as more proximally. Two forms of GLP-1 are secreted after a meal: GLP-1(7-37) and GLP-1(7-36) amide; these are similarly potent. Most of the circulating activity is due to GLP-1(7-36) amide. Release of GIP and GLP-1 by ingested food provides an early stimulus to insulin secretion before absorbed glucose or other products of digestion reach the islet cells in the portal blood (Fig. 30.1). As well as stimulating insulin secretion, both these hormones inhibit pancreatic glucagon secretion and slow the rate of absorption of digested food by reducing gastric emptying. They are also implicated in control of food intake via appetite and satiety (see Ch. 31). The actions of GIP and GLP-1 are terminated rapidly by dipeptidyl peptidase-4 (DPP-4). This enzyme is a membrane glycoprotein with rather wide substrate specificity—it has been implicated in suppression of malignancy (e.g. Wesley et al., 2005).
Diabetes Mellitus
Diabetes mellitus is a chronic metabolic disorder characterised by a high blood glucose concentration—hyper-glycaemia (fasting plasma glucose > 7.0 mmol/l, or plasma glucose > 11.1 mmol/l, 2 h after a meal)—caused by insulin deficiency, often combined with insulin resistance. Hyperglycaemia occurs because of uncontrolled hepatic glucose output and reduced uptake of glucose by skeletal muscle with reduced glycogen synthesis. When the renal threshold for glucose reabsorption is exceeded, glucose spills over into the urine (glycosuria) and causes an osmotic diuresis (polyuria) which, in turn, results in dehydration, thirst and increased drinking (polydipsia). Insulin deficiency causes wasting through increased breakdown and reduced synthesis of proteins. Diabetic ketoacidosis is an acute emergency. It develops in the absence of insulin because of accelerated breakdown of fat to acetyl-CoA, which, in the absence of aerobic carbohydrate metabolism, is converted to acetoacetate and β-hydroxybutyrate (which cause acidosis) and acetone (a ketone).
Various complications develop as a consequence of the metabolic derangements in diabetes, often over many years. Many of these are the result of disease of blood vessels, either large (macrovascular disease) or small (microangiopathy). Dysfunction of vascular endothelium (see Ch. 22) is an early and critical event in the development of vascular complications. Oxygen-derived free radicals, protein kinase C and non-enzymic products of glucose and albumin called advanced glycation end products (AGE) have been implicated. Macrovascular disease consists of accelerated atheroma (Ch. 23) and its thrombotic complications (Ch. 24), which are commoner and more severe in diabetic patients. Microangiopathy is a distinctive feature of diabetes mellitus and particularly affects the retina, kidney and peripheral nerves. Diabetes mellitus is the commonest cause of chronic renal failure, which itself represents a huge and rapidly increasing problem, the costs of which to society as well as to individual patients are staggering. Coexistent hypertension promotes progressive renal damage, and treatment of hypertension slows the progression of diabetic nephropathy and reduces the risk of myocardial infarction. Angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists (Ch. 22) are more effective in preventing diabetic nephropathy than other antihypertensive drugs, perhaps because they prevent fibroproliferative actions of angiotensin II and aldosterone.
Diabetic neuropathy5 is associated with accumulation of osmotically active metabolites of glucose, produced by the action of aldose reductase, but aldose reductase inhibitors have been disappointing as therapeutic drugs (see Chung & Chung, 2005, for a review).
There are two main types of diabetes mellitus:
In type 1 diabetes, there is an absolute deficiency of insulin resulting from autoimmune destruction of pancreatic B cells. Without insulin treatment, such patients will ultimately die with diabetic ketoacidosis.
Type 1 diabetic patients are usually young (children or adolescents) and not obese when they first develop symptoms. There is an inherited predisposition, with a 10-fold increased incidence in first-degree relatives of an index case, and strong associations with particular histocompatibility antigens (HLA types). Studies of identical twins have shown that genetically predisposed individuals must additionally be exposed to an environmental factor such as viral infection (e.g. with coxsackievirus or echovirus). Viral infection may damage pancreatic B cells and expose antigens that initiate a self-perpetuating autoimmune process. The patient becomes overtly diabetic only when more than 90% of the B cells have been destroyed. This natural history provides a tantalising prospect of intervening in the prediabetic stage, and a variety of strategies have been mooted, including immunosuppression, early insulin therapy, antioxidants, nicotinamide and many others; so far these have disappointed, but this remains a very active field.
Type 2 diabetes is accompanied both by insulin resistance (which precedes overt disease) and by impaired insulin secretion, each of which are important in its pathogenesis. Such patients are often obese and usually present in adult life, the incidence rising progressively with age as B-cell function declines. Treatment is initially dietary, although oral hypoglycaemic drugs usually become necessary, and about one-third of patients ultimately require insulin. Prospective studies have demonstrated a relentless deterioration in diabetic control6 over the years.
Insulin secretion in the two main forms of diabetes is shown schematically in Figure 30.2, contrasted with the normal response.
There are many other less common forms of diabetes mellitus in addition to the two main ones described above, and hyperglycaemia can also be a clinically important adverse effect of several drugs, including glucocorticoids (Ch. 32), high doses of thiazide diuretics (Ch. 28) and several of the protease inhibitors used to treat HIV infection (Ch. 51).
Treatment of Diabetes Mellitus
Insulin is essential for the treatment of type 1 diabetes, and a valuable component of the treatment of many patients with type 2 disease.
For many years, it was assumed, as an act of faith, that normalising plasma glucose would prevent diabetic complications. The Diabetes Control and Complications Trial (American Diabetes Association, 1993) showed that this faith was well placed: type 1 diabetic patients were randomly allocated to intensive or conventional management. Mean fasting blood glucose concentration was 2.8 mmol/l lower in the intensively treated group, who had a substantial reduction in the occurrence and progression of retinopathy, nephropathy and neuropathy over a period of 4–9 years. These benefits outweighed a three-fold increase in severe hypoglycaemic attacks and modest excess weight gain.
The UK Prospective Diabetes Study showed that lowering blood pressure markedly improves outcome in type 2 diabetes. Normalisation of blood glucose was not achieved even in intensively treated patients. Better metabolic control did improve outcome, but (in contrast to lowering blood pressure) the magnitude of the benefit was disappointing and statistically significant only for microvascular complications. In long-term follow-up, patients from this study who had been allocated to intensive treatment continued to have better outcomes than patients treated with diet alone (despite diabetic control becoming similar in the two groups after the blinded treatment period had finished), suggesting that early diabetic control (within the first 12 years from diagnosis) is important (Holman et al., 2008). By contrast, studies of intensive control later in the course of the disease have been disappointing with harm from hypoglycaemia outweighing any benefit of intensive treatment.
Realistic goals in type 2 diabetic patients are usually less ambitious than in younger type 1 patients. Diet is the cornerstone (albeit one with a tendency to crumble), combined with increased exercise. Oral agents are used to control symptoms from hyperglycaemia, as well as to limit microvascular complications, and are introduced early. Dietary measures and statins to prevent atheromatous disease (Ch. 24) are crucial. Details of dietary management and treatment for specific diabetic complications are beyond the scope of this book. Newer drugs (glitazones and drugs that mimic or potentiate incretins) have been shown to reduce glycated haemoglobin (typically by 0.5–1 percentage points) but their effects (if any) on clinical outcomes such as diabetic complications are unproven.
Insulin Treatment
The effects of insulin and its mechanism of action are described above. Here we describe pharmacokinetic aspects and adverse effects, both of which are central to its therapeutic use. Insulin for clinical use was once either porcine or bovine but is now almost entirely human (made by recombinant DNA technology). Animal insulins are liable to elicit an immune response, a problem that is avoided by the use of recombinant human insulin. Although recombinant insulin is more consistent in quality than insulins extracted from pancreases of freshly slaughtered animals, doses are still quantified in terms of units of activity, with which doctors and patients are familiar, rather than of mass.
Pharmacokinetic aspects and insulin preparations
Insulin is destroyed in the gastrointestinal tract, and must be given parenterally—usually subcutaneously, but intravenously or occasionally intramuscularly in emergencies. Intraperitoneal insulin is used in diabetic patients with end-stage renal failure treated by ambulatory peritoneal dialysis. Pulmonary absorption of insulin occurs, but an aerosol formulation was withdrawn from therapeutic use. Other potential approaches include incorporation of insulin into biodegradable polymer microspheres as a slow-release formulation, and its encapsulation with a lectin in a glucose-permeable membrane.7 Once absorbed, insulin has an elimination half-life of approximately 10 min. It is inactivated enzymically in the liver and kidney, and 10% is excreted in the urine. Renal impairment reduces insulin requirement.
One of the main problems in using insulin is to avoid wide fluctuations in plasma concentration and thus in blood glucose. Different formulations vary in the timing of their peak effect and duration of action. Soluble insulin produces a rapid and short-lived effect. Longer-acting preparations are made by precipitating insulin with protamine or zinc, thus forming finely divided amorphous solid or relatively insoluble crystals, which are injected as a suspension from which insulin is slowly absorbed. These preparations include isophane insulin and amorphous or crystalline insulin zinc suspensions. Mixtures of different forms in fixed proportions are available. Insulin lispro is an insulin analogue in which a lysine and a proline residue are ‘switched’. It acts more rapidly but for a shorter time than natural insulin, enabling patients to inject themselves immediately before the start of a meal. Insulin glargine is another modified insulin analogue, designed with the opposite intention, namely to provide a constant basal insulin supply and mimic physiological postabsorptive basal insulin secretion. Insulin glargine, which is a clear solution, forms a microprecipitate at the physiological pH of subcutaneous tissue, and absorption from the subcutaneous site of injection is prolonged. Used in conjunction with short-acting insulin, it lowers postabsorptive plasma glucose.
Various dosage regimens are used. Some type 1 patients inject a combination of short- and intermediate-acting insulins twice daily, before breakfast and before the evening meal. Improved control of blood glucose can be achieved with multiple daily injections of short-acting insulins with meals, and a longer-acting insulin at night. Insulin pumps are used in hospital and sometimes, by specialists, in outpatients. The most sophisticated forms of pump regulate the dose by means of a sensor that continuously measures blood glucose, but these are not routinely available.
Unwanted effects
The main undesirable effect of insulin is hypoglycaemia. This is common and, if very severe, can cause brain damage. In one large clinical trial, intensive insulin therapy resulted in a three-fold increase in severe hypoglycaemia compared with usual care. The treatment of hypoglycaemia is to take a sweet drink or snack or, if the patient is unconscious, to give intravenous glucose or intramuscular glucagon (see above). Rebound hyperglycaemia (‘Somogyi effect’) can follow insulin-induced hypoglycaemia, because of the release of counter-regulatory hormones (see above). This can cause hyperglycaemia before breakfast following an unrecognised hypoglycaemic attack during sleep in the early hours of the morning. It is essential to appreciate this possibility to avoid the mistake of increasing (rather than reducing) the evening dose of insulin in this situation.
Allergy to human insulin is unusual but can occur. It may take the form of local or systemic reactions. Insulin resistance as a consequence of antibody formation is rare. Theoretical concerns regarding mitogenic effects of insulin analogues are mentioned above.
Clinical uses of insulin and other hypoglycaemic drugs for injection 
Other Hypoglycaemic Agents
Biguanides
Metformin is the only drug of the biguanide class (originally found in French lilac, Galega officinalis) that is used clinically.
Actions and mechanism
Biguanides have several biochemical actions. They:
Reduced hepatic gluconeogenesis is especially important. The mechanism involves activation in hepatocytes of AMP-activated protein kinase (AMPK), an important enzyme in metabolic control (Towler & Hardie, 2007). Activation of AMPK increases expression of a nuclear receptor that inhibits expression of genes that are important for gluconeogenesis in the liver (see Kim et al., 2008 for details).
Metformin has a half-life of about 3 h and is excreted unchanged in the urine.
Unwanted effects
Metformin, while preventing hyperglycaemia, does not cause hypoglycaemia, and the commonest unwanted effects are dose-related gastrointestinal disturbances (e.g. anorexia, diarrhoea, nausea), which are usually but not always transient. Lactic acidosis is a rare but potentially fatal toxic effect, and metformin should not be given routinely to patients with renal or hepatic disease, hypoxic pulmonary disease or shock. Such patients are predisposed to lactic acidosis because of reduced drug elimination or reduced tissue oxygenation. Compensated heart failure is not a contraindication, and indeed metformin is associated with improved outcome in patients with diabetes and heart failure (Eurich et al., 2007). It should be avoided in other situations that predispose to lactic acidosis including some forms of mitochondrial myopathy that are associated with diabetes. Long-term use may interfere with absorption of vitamin B12.
Clinical use
Metformin is used to treat patients with type 2 diabetes. It does not stimulate appetite (rather the reverse; see above!) and is consequently the drug of first choice in the majority of type 2 patients who are obese, provided they have unimpaired renal and hepatic function. It can be combined with sulfonylureas, glitazones or insulin. Potential uses outside diabetes include other syndromes with accompanying insulin resistance including polycystic ovary syndrome, non-alcoholic fatty liver disease and some forms of premature puberty, but these indications remain experimental.
Sulfonylureas
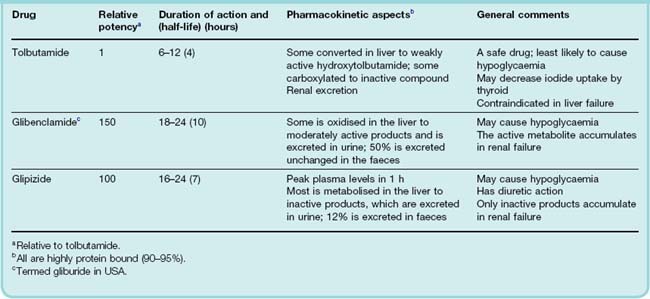
The sulfonylureas were developed following the chance observation that a sulfonamide derivative (used to treat typhoid) caused hypoglycaemia. Numerous sulfonylureas are available. The first used therapeutically were tolbutamide and chlorpropamide. Chlorpropamide has a long duration of action and a substantial fraction is excreted in the urine. Consequently, it can cause severe hypoglycaemia, especially in elderly patients in whom renal function declines inevitably but insidiously (Ch. 28). It causes flushing after alcohol because of a disulfiram-like effect (Ch. 48), and has an action like that of antidiuretic hormone on the distal nephron, giving rise to hyponatraemia and water intoxication. Williams (1994) comments that ‘time honoured but idiosyncratic chlorpropamide should now be laid to rest’—a sentiment with which we concur. Tolbutamide, however, remains useful. So-called second-generation sulfonylureas (e.g. glibenclamide, glipizide; see Table 30.3) are more potent (on a milligram basis), but their maximum hypoglycaemic effect is no greater and control of blood glucose no better than with tolbutamide. These drugs all contain the sulfonylurea moiety and act in the same way, but different substitutions result in differences in pharmacokinetics and hence in duration of action (see Table 30.3).
Mechanism of action
The principal action of sulfonylureas is on B cells (Fig. 30.1), stimulating insulin secretion and thus reducing plasma glucose. High-affinity receptors for sulfonylureas are present on the KATP channels (Ch. 4) in B-cell plasma membranes, and the binding of various sulfonylureas parallels their potency in stimulating insulin release. Block by sulfonylurea drugs of KATP channel activation causes depolarisation, Ca2+ entry and insulin secretion. (Compare this with the physiological control of insulin secretion, see above.)
Pharmacokinetic aspects
Sulfonylureas are well absorbed after oral administration, and most reach peak plasma concentrations within 2–4 h. The duration of action varies (Table 30.3). All bind strongly to plasma albumin and are implicated in interactions with other drugs (e.g. salicylates and sulfonamides) that compete for these binding sites (see below and Ch. 56). Most sulfonylureas (or their active metabolites) are excreted in the urine, so their action is increased in the elderly and in patients with renal disease.
Most sulfonylureas cross the placenta and enter breast milk and their use is contraindicated in pregnancy and in breastfeeding.
Unwanted effects
The sulfonylureas are usually well tolerated. Unwanted effects are specified in Table 30.3. The commonest adverse effect is hypoglycaemia, which can be severe and prolonged. Its incidence is related to the potency and duration of action of the agent, the highest incidence occurring with long-acting chlorpropamide and glibenclamide and the lowest with tolbutamide. Long-acting sulfonylureas are best avoided in the elderly and in patients with even mild renal impairment because of the risk of hypoglycaemia. Sulfonylureas stimulate appetite and often cause weight gain. This is a major concern in obese diabetic patients. About 3% of patients experience gastrointestinal upsets. Allergic skin rashes can occur, and bone marrow toxicity (Ch. 57), although rare, can be severe.
During and for a few days after acute myocardial infarction, insulin must be substituted for sulfonylurea treatment. This is associated with a substantial reduction in short-term mortality, although it remains unclear if this is due to a beneficial effect specific to insulin or to a detrimental effect of sulfonylurea drugs in this setting, or both. Another vexing question is whether prolonged therapy with oral hypoglycaemic drugs has adverse effects on the cardiovascular system. A study in the USA in the 1970s found that after 4–5 years of treatment, there was an increase in cardiovascular deaths in the group treated with oral drugs compared with the groups treated with insulin or placebo. Blockade of KATP in heart and vascular tissue could theoretically have adverse effects, but evidence for an adverse cardiovascular effect is inconclusive.
Drug interactions
Several drugs augment the hypoglycaemic effect of sulfonylureas. Non-steroidal anti-inflammatory drugs, coumarins, some uricosuric drugs (e.g. sulfinpyrazone), alcohol, monoamine oxidase inhibitors, some antibacterial drugs (including sulfonamides, trimethoprim and chloramphenicol) and some imidazole antifungal drugs have all been reported to produce severe hypoglycaemia when given with a sulfonylurea. The probable basis of most of these interactions is competition for metabolising enzymes, but interference with plasma protein binding or with transport mechanisms facilitating excretion may play some part.
Agents that decrease the action of sulfonylureas on blood glucose include high doses of thiazide diuretics and corticosteroids.
Other Drugs That Stimulate Insulin Secretion
Several drugs that act, like the sulfonylureas, by blocking the sulfonylurea receptor on KATP channels in pancreatic B cells but lack the sulfonylurea moiety have recently been developed. These include repaglinide and nateglinide which, though much less potent than most sulfonylureas, have rapid onset and offset kinetics leading to short duration of action and a low risk of hypoglycaemia.8 These drugs are administered shortly before a meal to reduce the postprandial rise in blood glucose in type 2 diabetic patients inadequately controlled with diet and exercise. They may cause less weight gain than conventional sulfonylureas. Later in the course of the disease, they can be combined with metformin or thiazolidinediones. Unlike glibenclamide, these drugs are relatively selective for KATP channels on B cells versus KATP channels in vascular smooth muscle.
Thiazolidinediones (glitazones)
The thiazolidinediones (or glitazones) were developed following the chance observation that a clofibrate analogue, ciglitazone, which was being screened for effects on lipids, unexpectedly lowered blood glucose. Ciglitazone caused liver toxicity, as did troglitazone, but there are only rare reports of hepatotoxicity with (rosiglitazone and pioglitazone which is the only drug of this class in clinical use.)
Effects
The effect of thiazolidinediones on blood glucose is slow in onset, the maximum effect being achieved only after 1–2 months of treatment. Thiazolidinediones:
They reduce the amount of exogenous insulin needed to maintain a given level of blood glucose by approximately 30%. Reduced blood glucose concentration is accompanied by reduced insulin and free fatty acid concentrations. Triglycerides decline, while LDL and high-density lipoprotein (HDL) are unchanged or slightly increased. The proportion of small dense LDL particles (believed to be the most atherogenic; Ch. 23) is reduced. Weight gain of 1–4 kg is common, usually stabilising in 6–12 months. Some of this is attributable to fluid retention: there is an increase in plasma volume of up to 500 ml, with a concomitant reduction in haemoglobin concentration caused by haemodilution; there is also an increase in extravascular fluid, and increased deposition of subcutaneous (as opposed to visceral) fat.
Mechanism of action
Thiazolidinediones bind to a nuclear receptor called the peroxisome proliferator-activated receptor-γ (PPARγ), which is complexed with retinoid X receptor (RXR; see Ch. 3).9 PPARγ occurs mainly in adipose tissue, but also in muscle and liver. It causes differentiation of adipocytes (this contributes to the unwanted effect of weight gain), increases lipogenesis and enhances uptake of fatty acids and glucose. It also promotes amiloride-sensitive sodium ion reabsorption in renal collecting ducts, explaining the adverse effect of fluid retention (Guan et al., 2005). Endogenous agonists of PPARγ include unsaturated fatty acids and various derivatives of these, including prostaglandin J2. Thiazolidinediones are exogenous agonists, which cause the PPARγ–RXR complex to bind to DNA, promoting transcription of several genes with products that are important in insulin signalling. These include lipoprotein lipase, fatty acid transporter protein, adipocyte fatty acid-binding protein, Glut-4, phosphoenolpyruvate carboxykinase, malic enzyme and others. It remains something of a mystery that glucose homeostasis should be so responsive to drugs that bind to receptors found mainly in fat cells; it has been suggested that the explanation may lie in resetting of the glucose–fatty acid (Randle) cycle by the reduction in circulating free fatty acids.
Pharmacokinetic aspects
Pioglitazone is rapidly and nearly completely absorbed, with time to peak plasma concentration of less than 2 h. It is highly (> 99%) bound to plasma proteins, and is subject to hepatic metabolism and has a short (< 7 h) elimination half-life for the parent drug, but substantially longer (up to 24 h) for the metabolite. Pioglitazone is metabolised mainly by a CYP2C isozyme and CYP3A4 to active metabolites, which are eliminated mainly in bile.
Unwanted effects
The serious hepatotoxicity of ciglitazone and troglitazone was not encountered during clinical trials of pioglitazone, and reports of liver dysfunction since general release have been rare. Regular blood tests of liver function are currently recommended. One (unproven) hypothesis is that the hepatotoxicity of troglitazone is caused by quinone metabolites of its α-tocopherol side chain. The commonest unwanted effects of pioglitazone are weight gain and fluid retention (see above). Fluid retention is a substantial concern, because it can precipitate or worsen heart failure, which contraindicates their use. In addition to increased cardiovascular risk, both observational studies and meta-analysis of randomised controlled trials (Loke et al., 2009) indicate an increased risk (approximately a doubling of risk) of fractures with chronic use. Symptoms of uncertain cause, including headache, fatigue and gastrointestinal disturbances, have also been reported. Thiazolidinediones are contraindicated in pregnant or breastfeeding women and in children. It is theoretically possible that these drugs could cause ovulation to resume in women who are anovulatory because of insulin resistance (e.g. with polycystic ovary syndrome). Another glitazone (rosiglitazone) was withdrawn from clinical use recently because of concerns over excess cardiovascular risks.
Clinical use
Because insulin resistance is one important component of the pathogenesis of type 2 diabetes, and has been implicated in the excess cardiovascular mortality that accompanies the common ‘metabolic syndrome’ (visceral obesity, hypertension, dyslipidaemia, insulin resistance, etc.), there is a good rationale for pioglitazone in type 2 diabetes. There is, however, as yet no evidence that this optimism is justified in terms of improved clinical outcomes (see, for example, Gale, 2001)—cardiovascular clinical end-point trials to rebut this view are still awaited. Pioglitazone is additive with other oral hypoglycaemic drugs in terms of effect on blood glucose, and short-term studies support their use in combination with metformin or with a sulfonylurea in patients whose blood glucose is inadequately controlled on one of these drugs and are unsuited to addition of the other.
α-Glucosidase inhibitors
Acarbose, an inhibitor of intestinal α-glucosidase, is used in type 2 patients whose diabetes is inadequately controlled by diet with or without other agents. It delays carbohydrate absorption, reducing the postprandial increase in blood glucose. The commonest adverse effects are related to its main action and consist of flatulence, loose stools or diarrhoea, and abdominal pain and bloating. Like metformin, it may be particularly helpful in obese type 2 patients, and it can be co-administered with metformin.
Incretin mimetics and related drugs
Exenatide is a synthetic version of exendin-4, a peptide found in the saliva of the Gila monster (a lizard, which presumably evolved this as means to disable its prey by rendering them hypoglycaemic).
Exenatide mimics the effects of GLP-1 (see above), but is longer acting. It lowers blood glucose after a meal by increasing insulin secretion, suppressing glucagon secretion and slowing gastric emptying (see above). It reduces food intake (by an effect on satiety) and is associated with modest weight loss. It reduces hepatic fat accumulation.
Exenatide is not absorbed by the gut and is administered subcutaneously. It is much more stable than GLP-1, and is administered twice daily before the first and last meal of the day. A long-acting preparation (for once-weekly administration) is under investigation (Drucker et al., 2008). It can cause hypoglycaemia and a range of gastrointestinal effects. Pancreatitis is a rare but sometimes severe problem.
Exenatide is used in patients with type 2 diabetes in combination with metformin with or without a sulfonylurea when these have been inadequate.
Gliptins
Gliptins (e.g. sitagliptin, vildagliptin) are synthetic drugs that competitively inhibit dipeptidylpeptidase-4 (DPP-4), thereby lowering blood glucose by potentiating endogenous incretins (GLP-1 and GIP, see above). Sitagliptin does not cause weight loss or weight gain.
Sitagliptin is well absorbed from the gut and is administered once daily by mouth. It is mainly eliminated by renal excretion and is also metabolised by hepatic CYP enzymes. It is well tolerated with an adverse effect profile in clinical trials similar to placebo, and similar occurrence of hypoglycaemia between placebo and sitagliptin. Vildagliptin is not available in the USA, where the Food and Drug Administration has required further investigation to exclude skin and renal toxicity.
Sitagliptin is used for type 2 diabetes, usually in addition to other oral hypoglycaemic drugs (see clinical box on uses of oral hypoglycaemic drugs).
Clinical uses of oral hypoglycaemic drugs
Potential New Antidiabetic Drugs
Several agents are currently being studied, including α2 adrenoceptor antagonists, inhibitors of fatty acid oxidation and activators of glucokinase. Lipolysis in fat cells is controlled by adrenoceptors of the β3 subtype (see Ch. 14). The possibility of using selective β3 agonists, currently in development, in the treatment of obese patients with type 2 diabetes is being investigated (see Ch. 31). There is interest in inhibitors of protein kinase C, for example ruboxistaurin, an inhibitor specific for the β isoform of protein kinase C, because of evidence implicating activation of this pathway in the development of vascular diabetic complications (Aiello, 2005)—a clinical trial is ongoing.
Drugs used in diabetes mellitus
Insulin and other injectable drugs
Oral hypoglycaemic drugs
References and Further Reading
Drucker D.J., Buse J.B., Taylor K., et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes. A randomised, open-label, non-inferiority study. Lancet. 2008;372:1240-1250.
Holman R.R., Sanjoy K.P., Bethel M.A., et al. 10-Year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med.. 2008;359:1577-1589.
Kim Y.D., Park K.G., Lee Y.S., et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes. 2008;57:306-314.
Loke Y.K., Singh S., Furberg C.D., et al. Long-term use of thiazolidinediones and fractures in type 2 diabetes: a meta-analysis. CMAJ. 2009;180:32-39. (Long-term thiazolidinedione use doubles the risk of fractures among women with type 2 diabetes, without a significant increase in risk of fractures among men with type 2 diabetes)
Towler M.C., Hardie D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res.. 2007;100:328-341.
Wesley U.V., McGroarty M., Homoyouni A. Dipeptidyl peptidase inhibits malignant phenotype of prostate cancer cells by blocking basic fibroblast growth factor signaling pathway. Cancer Res.. 2005;65:1325-1334.
Physiological and pathophysiological aspects
Lee Y.H., White M.F. Insulin receptor substrate proteins and diabetes. Arch. Pharm. Res.. 2004;27:361-370. (Reviews the discovery of IRS proteins and their role linking cell surface receptors to intracellular signalling cascades. ‘Understanding the regulation and signaling by IRS1 and IRS2 in cell growth, metabolism and survival will reveal new strategies to prevent or cure diabetes and other metabolic diseases.’)
Withers D.J., Gutierrez J.S., Towery H., et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900-904. (Dysfunction of IRS-2 may ‘contribute to the pathophysiology of human type 2 diabetes’; see also accompanying commentary by Avruch, J., A signal for β-cell failure, pp. 846–847)
Zimmet P., Alberti K.G.M.M., Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782-787. (Changes in human behaviour have resulted in a dramatic increase in type 2 diabetes worldwide)
Owens D.R., Zinman B., Bolli G.B. Insulins today and beyond. Lancet. 2001;358:739-746. (Reviews the physiology of glucose homeostasis, genetically engineered ‘designer’ insulins and developments in insulin delivery and glucose sensing)
Eurich D.T., McAlister F.A., Blackburn D.F., et al. Benefits and harms of antidiabetic agents in patients with diabetes and heart failure: systematic review. BMJ. 2007;335:497-507. (Metformin was the only agent not associated with harm in patients with diabetes and heart failure)
Gale E.A.M. Lessons from the glitazones: a story of drug development. Lancet. 2001;357:1870-1875. (Fighting stuff: ‘Troglitazone was voluntarily withdrawn in Europe, but went on to generate sales of over $2 billion in the USA and caused 90 cases of liver failure before being withdrawn. Rosiglitazone and pioglitazone reached the USA for use alone or in combination with other drugs whereas in Europe the same dossiers were used to apply for a limited licence as second-line agents. How should we use them? How did they achieve blockbuster status without any clear evidence of advantage over existing therapy?’)
Guan Y., Hao C., Cha D.R., et al. Thiazolidinediones expand body fluid volume through PPARγ stimulation of ENaC-mediated renal salt absorption. Nat. Med.. 2005;11:861-865. (Mechanism of fluid retention caused by thiazolidinediones and suggestion that amiloride may provide a specific therapy for this. Human studies will no doubt follow … See also News and Views article in the same issue: TZDs and diabetes: testing the waters, by A.F. Semenkovich, pp. 822–824)
Other drugs for diabetes, and therapeutic aspects
Brenner B.M., Cooper M.E., de Zeeuw D., et al Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy N. Engl. J. Med. 345:2001:861-869 (Significant renal benefits from the AT1 antagonist; see also two adjacent articles: Lewis, E.J., et al., pp. 851–860, and Parving, H.-H., et al., pp. 870–878, and an editorial on prevention of renal disease caused by type 2 diabetes by Hostetter, T.H., pp. 910–911)
Chung S.S.M., Chung S.K. Aldose reductase in diabetic microvascular complications. Curr. Drug Targets. 2005;6:475-486. (Reviews pathogenic mechanisms of the polyol pathway, and discusses possible reasons for the unimpressive effects to date of aldose reductase inhibitors; argues that renewed efforts could be warranted)
Schmitz O., Brock B., Rungby G. Amylin agonists: a novel approach in the treatment of diabetes. Diabetes. 2004;53(Suppl.):S233-S238. (Reviews actions of amylin in animal and human models, and the results from clinical trials with the amylin analogue pramlintide)
1Not to be confused with C-reactive peptide, which is an acute-phase reactant used clinically as a marker of inflammation (Ch. 6).
2Insulin for injection does not contain C-peptide, which therefore provides a means of distinguishing endogenous from exogenous insulin. This is used to differentiate insulinoma (an insulin-secreting tumour causing high circulating insulin with high C-peptide) from surreptitious injection of insulin (high insulin, normal or low C-peptide). Deliberate induction of hypoglycaemia by self-injection with insulin is a well-recognised, if unusual, manifestation of psychiatric disorder, especially in health professionals—it has also been used in murder.
3The action on K+ is exploited in the emergency treatment of hyperkalaemia by intravenous glucose with insulin (see Ch. 28).
4Octreotide is used either short term before surgery on the pituitary tumour, or while waiting for radiotherapy of the tumour to take effect, or if other treatments have been ineffective.
5Neuropathy (’disease of the nerves’) causes dysfunction of peripheral nerve fibres, which can be motor, sensory or autonomic. Diabetic neuropathy often causes numbness in a ‘stocking’ distribution caused by damage to sensory fibres, and postural hypotension and erectile dysfunction due to autonomic neuropathy.
6Diabetic control is not easily estimated by determination of blood glucose, because this is so variable. Instead, glycated haemoglobin (haemoglobin A1C) is measured. This provides an integrated measure of control over the lifespan of the red cell: approximately 120 days.
7This could, in theory, provide variable release of insulin controlled by the prevailing glucose concentration, because glucose and glycated insulin compete for binding sites on the lectin.
8It is ironic that these aggressively marketed drugs share many of the properties of tolbutamide, the oldest, least expensive and least fashionable of the sulfonylureas. Perhaps diabetologists should turn some of their investigative effort to studying how best to use this Cinderella drug!
9Compare with fibrates (to which thiazolidinediones are structurally related), which bind to PPARα (see Ch. 23).