Chronic kidney disease (CKD)
The term ‘CKD’ has replaced terms such as chronic renal failure or insufficiency. CKD implies longstanding (more than 3 months), and usually progressive, impairment in renal function. In many instances, no effective means are available to reverse the primary disease process. Exceptions include correction of urinary tract obstruction, immunosuppressive therapy for systemic vasculitis and Goodpasture’s syndrome, treatment of accelerated hypertension, and correction of critical narrowing of renal arteries causing CKD. The rate of deterioration in renal function can, however, be slowed (see p. 622). A list of causes of CKD is given in Table 12.21.
Table 12.21 Causes of chronic kidney disease
Wide geographical variations in the incidence of disorders causing CKD exist. The most common cause of glomerulonephritis in sub-Saharan Africa is malaria. Schistosomiasis is a common cause of CKD due to urinary tract obstruction in parts of the Middle East, including southern Iraq. The incidence of ESKD varies between racial groups. ESKD is three to four times as common in black Africans in the UK and USA as it is in whites, and hypertensive nephropathy is a much more frequent cause of ESKD in this group. The prevalence of diabetes mellitus and hence of diabetic nephropathy is higher in some Asian groups than in whites. The age is of relevance; CKD due to atherosclerotic renal vascular disease is much more common in the elderly than in the young. Over 70% of all cases with CKD are due to diabetes mellitus, hypertension and atherosclerosis.
Patients with CKD should be referred to a nephrologist with access to facilities for renal replacement therapy at an early stage since late referral has been shown to be associated with increased mortality and morbidity when such patients commence renal replacement therapy. Old age is no bar to referral in the reasonably fit elderly patient.
For CKD referral and treatment pathways, the Kidney Disease Outcomes Quality Initiative (K/DOQI) of the National Kidney Foundation established a classification of CKD in the USA in 2002, and endorsed by the Kidney Disease: Improving Global Outcomes (KDIGO) in 2004. According to KDIGO and KDOQI, the definition and classification should reflect patient prognosis and outcomes. KDIGO initiated a collaborative meta-analysis to examine the relationship of eGFR and albuminuria to mortality and kidney outcomes. It has been agreed by experts that:
 The current definition for CKD will be retained: GFR <60 mL/min per 1.73 m2 or a urinary albumin-to creatinine ratio >65 mg/mmol or protein creatinine ratio of 100 mg/mmol (Table 12.22)
The current definition for CKD will be retained: GFR <60 mL/min per 1.73 m2 or a urinary albumin-to creatinine ratio >65 mg/mmol or protein creatinine ratio of 100 mg/mmol (Table 12.22)
The classification has been modified by adding albuminuria stage, subdivision of stage 3 into A and B, and emphasizing clinical diagnosis.
Table 12.22 Classification of CKD
| Stage | GFR (mL/min/1.73 m2) | Description |
|---|---|---|
1 |
≥90 |
Normal or increased glomerular filtration rate (GFR), with other evidence of kidney damage |
2 |
60–89 |
Slight decrease in GFR with other evidence of kidney damage |
3A |
45–59 |
Moderate decrease in GFR with or without other evidence of kidney damage |
3B |
30–44 |
|
4 |
15–29 |
Severe decrease in GFR with or without other evidence of kidney damage |
5 |
<15 |
Established renal failure |
Note: The suffix ‘P’ can be applied to the stage of CKD if the patient has significant proteinuria, defined as a urinary albumin:creatinine ratio >65 mg/mmol or protein:creatinine ratio >100 mg/mmol.
Source: NICE Clinical guidance 73. Chronic kidney disease. 2011.
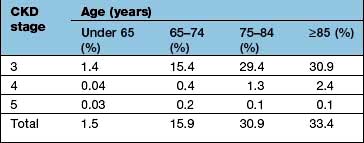
High prevalences of CKD have now been found in population-based surveys particularly in elderly patients (Table 12.23) using the MDRD method (p. 565) to estimate GFR. Adoption of albuminuria or proteinuria along with eGFR in the new proposed classification will, according to some estimates, reduce the prevalence of CKD stages 3 and 4 from 10–15% of total population to more realistic numbers of less than 5% and would in the process avoid unnecessary referrals.
FURTHER READING
Levey AS, de Jong PE, Coresh J et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011; 80(1):17–28.
Tonelli M, Muntner P, Lloyd A et al. Using proteinuria and estimated glomerular filtration rate to classify risk in patients with chronic kidney disease: a cohort study. Ann Intern Med 2011; 154(1):12–21.
Clinical approach to the patient with CKD or any other form of renal disease
History
Particular attention should be paid to:
Drug ingestion, including non-steroidal anti-inflammatory agents, analgesic and other medications, and unorthodox treatments such as herbal remedies
Previous medical and surgical history, e.g. previous chemotherapy, multisystem diseases such as SLE, malaria
Previous occasions on which urinalysis or measurement of urea and creatinine might have been performed, e.g. pre-employment or insurance medical examinations, new patient checks
Symptoms
The early stages of CKD are often completely asymptomatic, despite the accumulation of numerous metabolites. Serum urea and creatinine concentrations are measured in CKD, since methods for their determination are available and a rough correlation exists between urea and creatinine concentrations and symptoms. These substances are, however, in themselves not particularly toxic. The nature of the metabolites that are involved in the genesis of symptoms is unclear. Such metabolites must be products of protein catabolism (since dietary protein restriction may reverse symptoms associated with CKD) and many of them must be of relatively small molecular size (since haemodialysis employing membranes which allow through only relatively small molecules improves symptoms). Little else is known with certainty.
Symptoms are common when the serum urea concentration exceeds 40 mmol/L, but many patients develop uraemic symptoms at lower levels of serum urea. Symptoms include:
Nocturia and polyuria due to impaired concentrating ability
Nausea, vomiting and diarrhoea
Paraesthesiae due to polyneuropathy
‘Restless legs’ syndrome (overwhelming need to frequently alter position of lower limbs)
Bone pain due to metabolic bone disease
Paraesthesiae and tetany due to hypocalcaemia
Symptoms due to salt and water retention – peripheral or pulmonary oedema
In more advanced uraemia CKD stage 5, these symptoms become more severe and CNS symptoms are common:
Severe depression of glomerular filtration can result in oliguria. This can occur with either acute kidney injury or in the terminal stages of CKD. However, even if the GFR is profoundly depressed, failure of tubular reabsorption may lead to very high urine volumes; the urine output is therefore not a useful guide to renal function.
Examination
There are few physical signs of uraemia per se. Findings include: short stature (in patients who have had CKD in childhood); pallor (due to anaemia); increased photosensitive pigmentation (which may make the patient look misleadingly healthy); brown discoloration of the nails; scratch marks due to uraemic pruritus; signs of fluid overload (p. 642); pericardial friction rub; flow murmurs (mitral regurgitation due to mitral annular calcification; aortic and pulmonary regurgitant murmurs due to volume overload); and glove and stocking peripheral sensory loss (rare).
The kidneys themselves are usually impalpable unless grossly enlarged as a result of polycystic disease, obstruction or tumour. Rectal and vaginal examination may disclose evidence of an underlying cause of CKD, particularly urinary obstruction, and should always be performed.
In addition to these findings, there may be physical signs of any underlying disease which may have caused the CKD, for instance:
Cutaneous vasculitic lesions in systemic vasculitides
Retinopathy in diabetes and hypertensive retinopathy in hypertension
Evidence of peripheral vascular disease and associated renal artery stenosis
Evidence of spina bifida or other causes of neurogenic bladder.
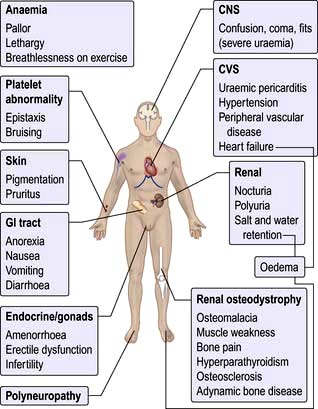
An assessment of the central venous pressure, skin turgor, blood pressure both lying and standing and peripheral circulation should also be made. The major symptoms and signs of CKD are shown in Figure 12.45.
Investigations
The following investigations are common for all renal patients. This includes patients with glomerular or non-glomerular disease, renal involvement in systemic diseases, AKI and CKD, as renal symptoms and signs are nonspecific.
Urinalysis
Haematuria may indicate glomerulonephritis, but other sources must be excluded. Haematuria should not be assumed to be due to the presence of an indwelling catheter.
Proteinuria, if heavy, is strongly suggestive of glomerular disease. Urinary infection may also cause proteinuria. Glycosuria with normal blood glucose is common in CKD.
Urine culture, including early-morning urine samples for TB.
Urine microscopy (see p. 569)
White cells in the urine usually indicate active bacterial urinary infection, but this is an uncommon cause of CKD; sterile pyuria suggests papillary necrosis or renal tuberculosis.
Eosinophiluria is strongly suggestive of allergic tubulointerstitial nephritis or cholesterol embolization.
Casts. Granular casts are formed from abnormal cells within the tubular lumen, and indicate active renal disease. Red-cell casts are highly suggestive of glomerulonephritis.
Red cells in the urine may be from anywhere between the glomerulus and the urethral meatus (Fig. 12.6).
Urine biochemistry
Measurements of urinary electrolytes are unhelpful in CKD. The use of urinary sodium concentration in the distinction between pre-renal and intrinsic renal disease is discussed on page 576.
Urine osmolality is a measure of concentrating ability. A low urine osmolality is normal in the presence of a high fluid intake but indicates renal disease when the kidney should be concentrating urine, such as in hypovolaemia or hypotension.
Urine electrophoresis and immunofixation is necessary for the detection of light chains, which can be present without a detectable serum paraprotein.
Haematology
Eosinophilia suggests vasculitis, allergic tubulointerstitial nephritis, or cholesterol embolism.
Markedly raised viscosity or ESR suggests myeloma or vasculitis.
Fragmented red cells and/or thrombocytopenia suggest intravascular haemolysis due to accelerated hypertension, haemolytic uraemic syndrome or thrombotic thrombocytopenic purpura.
Tests for sickle cell disease should be performed when relevant.
Immunology
Complement components may be low in active renal disease due to SLE, mesangiocapillary glomerulonephritis, post-streptococcal glomerulonephritis, and cryoglobulinaemia.
Autoantibody screening is useful in detection of SLE (p. 537); scleroderma (p. 539); Wegener’s granulomatosis and microscopic polyangiitis (p. 854); and Goodpasture’s syndrome (p. 812).
Cryoglobulins in unexplained glomerular disease, particularly mesangiocapillary glomerulonephritis.
Antibodies to streptococcal antigens (antistreptolysin O titre (ASOT), anti-DNAse B) if post-streptococcal glomerulonephritis is possible.
Antibodies to hepatitis B and C may point to polyarteritis or membranous nephropathy (hepatitis B) or to cryoglobulinaemic renal disease (hepatitis C).
Antibodies to HIV raise the possibility of HIV-associated renal disease.
Radiological investigation
Ultrasound. Every patient should undergo ultrasonography (for renal size and to exclude hydronephrosis), and plain abdominal radiography and CT (without contrast) to exclude low-density renal stones or nephrocalcinosis, which may be missed on ultrasound.
CT is also useful for the diagnosis of retroperitoneal fibrosis and some other causes of urinary obstruction, and may also demonstrate cortical scarring.
MRI. Magnetic resonance angiography in renovascular disease. For gadolinium used as contrast in CKD, see this chapter.
Renal biopsy (see p. 572)
This should be performed in every person with unexplained CKD and normal-sized kidneys, unless there are strong contraindications. If rapidly progressive glomerulonephritis is possible, this investigation must be performed within 24 h of presentation if at all possible.
Complications of chronic kidney disease
Anaemia
Several factors have been implicated:
Erythropoietin deficiency (the most significant)
Bone marrow toxins retained in CKD
Bone marrow fibrosis secondary to hyperparathyroidism
Haematinic deficiency – iron, vitamin B12, folate
Increased red-cell destruction
Abnormal red-cell membranes causing increased osmotic fragility
Increased blood loss – occult gastrointestinal bleeding, blood sampling, blood loss during haemodialysis or because of platelet dysfunction
ACE inhibitors (may cause anaemia in CKD, probably by interfering with the control of endogenous erythropoietin release).
Red-cell survival is reduced in CKD. Increased red-cell destruction may occur during haemodialysis owing to mechanical, oxidant and thermal damage.
Bone disease: renal osteodystrophy
The term ‘renal osteodystrophy’, more appropriately described as bone mineral disorder, embraces the various forms of bone disease that may develop alone or in combination in CKD – hyperparathyroid bone disease, osteomalacia, osteoporosis, osteosclerosis and adynamic bone disease (Fig. 12.46). Most patients with CKD are found, histologically, to have mixed bone disease. Covert renal osteodystrophy is present in many patients with moderate CKD and in almost all of those with ESKD.
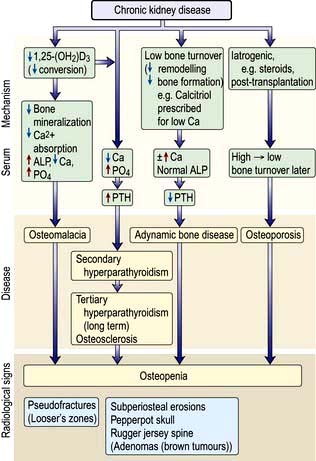
Figure 12.46 Renal osteodystrophy: pathogenesis and radiological features of renal bone disease. ALP, alkaline phosphatase.
Pathogenesis of bone disease
Phosphate retention owing to reduced excretion by the kidneys occurs in the very early stages of CKD. This results in the release of fibroblast growth factor 23 (FGF 23) and other phosphaturic agents by osteoblasts as a compensatory mechanism. FGF 23 causes phosphaturia to bring the plasma phosphate level to within the normal range. FGF 23 also downregulates 1α-hydroxylase in an attempt to reduce intestinal absorption of phosphate. However, consistently elevated levels of FGF 23 after a while cannot control phosphate levels and its effects are overwhelmed by development of secondary hyperparathyroidism. Elevated FGF 23 levels are the strongest independent predictor of mortality in patients with CKD. This underscores the necessity of controlling phosphate levels during very early stages of CKD.
Decreased renal production of the 1α-hydroxylase enzyme results in reduced conversion of 25-(OH)2D3 to the more metabolically active 1,25-(OH)2D3.
Reduced activation of vitamin D receptors (VDR) in the parathyroid glands leads to increased release of parathyroid hormone (secondary hyperparathyroidism).
The calcium sensing receptors (CaR), expressed in the parathyroid glands, react rapidly to acute changes in extracellular calcium concentrations and a low calcium also leads to increased release of PTH.
1,25-Dihydroxycholecalciferol deficiency also results in gut calcium malabsorption.
Phosphate retention owing to reduced excretion by the kidneys, also indirectly by lowering ionized calcium (and probably directly via a putative but unrecognized phosphate receptor), results in an increase in PTH synthesis and release.
PTH promotes reabsorption of calcium from bone and increased proximal renal tubular reabsorption of calcium, and this opposes the tendency to develop hypocalcaemia induced by 1,25-(OH)2D3 deficiency and phosphate retention. This ‘secondary’ hyperparathyroidism leads to increased osteoclastic activity, cyst formation and bone marrow fibrosis (osteitis fibrosa cystica).
Radiologically, digital subperiosteal erosions and ‘pepperpot skull’ are seen. Longstanding secondary hyperparathyroidism ultimately leads to hyperplasia of the glands with autonomous or tertiary hyperparathyroidism in which hypercalcaemia is present. Serum alkaline phosphatase concentration is raised in both secondary and tertiary hyperparathyroidism. Longstanding parathyroid hormone excess is also thought to cause increased bone density (osteosclerosis) seen particularly in the spine where alternating bands of sclerotic and porotic bone give rise to a characteristic ‘rugger jersey’ appearance on X-ray.
1,25-(OH)2D3 deficiency and hypocalcaemia result in impaired mineralization of osteoid (osteomalacia). Such impaired mineralization also occurs when osteoblasts are inhibited by, e.g. aluminium given as gut phosphorus binders, or accumulated in bone as a result of exposure to aluminium in source water used to make up dialysate for haemodialysis. In this situation, serum alkaline phosphatase concentration tends to be low or normal.
The condition of ‘adynamic bone disease’ in which both bone formation and resorption are depressed (in the absence of aluminium bone disease or overtreatment with vitamin D) is also seen. The pathogenesis of this condition is unclear and it is not known whether it leads to an increased risk of fractures or other complications. There may be hypercalcaemia; the serum alkaline phosphatase is normal, the PTH is low. X-rays and dual X-ray absorptiometry (DXA) scan show osteopenia. No treatment is of proven benefit.
Osteoporosis is commonly found in CKD, often after transplantation and the use of corticosteroids. Monitoring is with yearly DXA scan.
Management is discussed on page 555.
Skin disease
Pruritus (itching) is common in severe CKD and is mainly due to retention of nitrogenous waste products of protein catabolism as it improves following dialysis. Other causes of pruritus include hypercalcaemia, hyperphosphataemia, elevated calcium × phosphate product, hyperparathyroidism (even if calcium and phosphate levels are normal) and iron deficiency.
In dialysis patients, inadequate dialysis is usually the cause of pruritus. Nevertheless, a significant number of dialysis patients who are well dialysed and in whom other causes of pruritus can be excluded suffer persistent itching. The cause is unknown and no effective treatment exists.
Many patients with CKD suffer from dry skin for which simple aqueous creams are helpful. Eczematous lesions, particularly in relation to the region of an arteriovenous fistula, are relatively common. CKD may also cause porphyria cutanea tarda (PCT), a blistering photosensitive skin rash. This results from a decrease in hepatic uroporphyrinogen decarboxylase combined with a decreased clearance of porphyrins in the urine or by dialysis. Pseudoporphyria, a condition similar to PCT but without enzyme deficiency, is also seen in CKD with increased frequency.
Nephrogenic systemic fibrosis (NSF)
NSF is a systemic fibrosing disorder with predominant skin involvement. It is seen only in patients with moderate to severe CKD (eGFR <30 mL/min), particularly patients on dialysis. Gadolinium-containing contrast agents, which are excreted exclusively by the kidney, have been implicated in the causation of over 95% cases of NSF (see p. 1220).
The diagnosis is based upon a biopsy of an involved site, showing proliferation of dermal fibrocytes with excessive collagen deposition. Special testing may show gadolinium.
NSF usually follows a chronic and unremitting course, with 30% having no improvement, 20% having modest improvement and 30% dying. No single therapy or combination of therapies has shown consistent benefit in NSF with exception of improvement in renal function. Improvement in the NSF may follow renal transplantation. Prevention is by avoiding the use of gadolinium-based contrast agents in patents with severe CKD (eGFR <30 mL/min) or those on dialysis therapy.
Gastrointestinal complications
These include decreased gastric emptying and increased risk of reflux oesophagitis, peptic ulceration, acute pancreatitis and constipation, particularly in patients on continuous ambulatory peritoneal dialysis (CAPD).
Elevations of serum amylase of up to three times normal may be found in CKD without any evidence of pancreatic disease, owing to retention of high-molecular-weight forms of amylase normally excreted in the urine.
Metabolic abnormalities
Gout. Urate retention is a common feature of CKD. Treatment of clinical gout is complicated by the nephrotoxic potential of NSAIDs. Colchicine is useful for the acute attack, and allopurinol should be introduced under colchicine cover to prevent further attacks. The dose of allopurinol should be reduced in CKD, e.g. 100 mg on alternate days.
Insulin. Insulin is catabolized by and to some extent excreted via the kidneys. For this reason, insulin requirements in diabetic patients decrease as CKD progresses. By contrast, end-organ resistance to insulin is a feature of advanced CKD resulting in modestly impaired glucose tolerance. Insulin resistance may contribute to hypertension and lipid abnormalities.
Lipid metabolism abnormalities. These are common in CKD, and include:
The situation is further complicated in ESKD, when regular heparinization (in haemodialysis), excessive glucose absorption (in CAPD) and immunosuppressive drugs (in transplantation) may all contribute to lipid abnormalities. Correction of lipid abnormalities by, e.g. HMG-CoA reductase inhibitor therapy (statins), is used in patients with CKD, although without formal proof of survival benefit.
Endocrine abnormalities
Hyperprolactinaemia, which may present with galactorrhoea in men as well as women
Increased luteinizing hormone (LH) levels in both sexes, and abnormal pulsatility of LH release
Decreased serum testosterone levels (only seldom below the normal level). Erectile dysfunction and decreased spermatogenesis are common
Absence of normal cyclical changes in female sex hormones, resulting in oligomenorrhoea or amenorrhoea
Complex abnormalities of growth hormone secretion and action, resulting in impaired growth in uraemic children (pharmacological treatment with recombinant growth hormone and insulin-like growth factor is used)
Abnormal thyroid hormone levels, partly because of altered protein binding. Measurement of thyroid-stimulating hormone (TSH) is the best way to assess thyroid function. True hypothyroidism occurs with increased frequency in CKD.
Muscle dysfunction
Uraemia appears to interfere with muscle energy metabolism, but the mechanism is uncertain. Decreased physical fitness (cardiovascular deconditioning) also contributes.
Nervous system
Central nervous system
Severe uraemia causes an unusual combination of depressed cerebral function and decreased seizure threshold. However, convulsions in a uraemic patient are much more commonly due to other causes such as accelerated hypertension, thrombotic thrombocytopenic purpura or drug accumulation. Asterixis, tremor and myoclonus are also features of severe uraemia.
Rapid correction of severe uraemia by haemodialysis leads to dialysis disequilibrium owing to osmotic cerebral swelling. This can be avoided by correcting uraemia gradually by short, repeated haemodialysis treatments or by the use of peritoneal dialysis.
‘Dialysis dementia’ is a syndrome of progressive intellectual deterioration, speech disturbance, myoclonus and fits, which is due to aluminium intoxication; it may be accompanied by aluminium bone disease and by microcytic anaemia. Low-grade aluminium exposure may also cause more subtle, subclinical deterioration in intellectual function. Prevention involves removal of aluminium from source water used to manufacture dialysis fluid, and restriction or avoidance of aluminium-containing gut phosphorus binders. Treatment is with the chelating agent desferrioxamine.
Psychiatric problems are common. Patients can have anxiety, depression, phobias and psychoses.
Autonomic nervous system
Increased circulating catecholamine levels associated with downregulation of α-receptors, impaired baroreceptor sensitivity and impaired efferent vagal function are common in CKD.
Overactivity of the sympathetic nervous system in CKD is believed to play a part in the genesis of hypertension in this condition. All of these abnormalities improve to some extent after institution of regular dialysis and resolve after successful renal transplantation.
Peripheral nervous system
Median nerve compression in the carpal tunnel is common, usually due to β2-microglobulin-related amyloidosis. This can be avoided by haemofiltration and haemodiafiltration.
‘Restless legs’ syndrome (p. 620) is common in uraemia. The syndrome is difficult to treat. Iron deficiency should be treated if present. Attention should be paid to adequacy of dialysis. Symptoms may improve with the correction of anaemia by erythropoietin. Clonazepam is sometimes useful. Renal transplantation cures the problem.
A polyneuropathy occurs in patients who are inadequately dialysed.
Calciphylaxis
Also known as calcific uraemic arteriolopathy, this is a rare but serious life-threatening complication in CKD patients. It is increasingly recognized as a contributing factor to death in dialysis patients. Aetiological factors include reduced serum levels of a calcification inhibitory protein (fetuin-A) and abnormalities in smooth muscle cell biology in uraemia. It presents as painful non-healing eschars with panniculitis and dermal necrosis. The characteristic feature on histology is vascular calcification and superimposed small vessel thrombosis (Fig. 12.47).
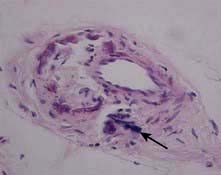
Figure 12.47 Calciphylaxis: characteristic arterial calcification of the skin microvasculature in the absence of vasculitic change (arrow).
Hyperparathyroidism and elevated concentrations of serum phosphate, morbid obesity and warfarin use remain consistent clinical features of most cases reported. Control of hyperparathyroidism is with either surgical intervention or with a calcimimetic agent.
Promising treatment options include hyperbaric oxygen therapy and sodium thiosulfate infusion. Benefits from bisphosphonates and tissue plasminogen activator have also been reported.
Cardiovascular disease
Life expectancy remains severely reduced compared with the normal population owing to a greatly increased (16-fold) incidence of cardiovascular disease, particularly myocardial infarction, cardiac failure, sudden cardiac death and stroke.
Risk factors
Hypertension is a frequent complication of CKD. Diabetes mellitus is the commonest cause of CKD. Dyslipidaemia is universal in uraemic patients. Furthermore, smoking is as common as in the general population and male gender is over-represented in patients with CKD. Ventricular hypertrophy is common, as is systolic and diastolic dysfunction. Diastolic dysfunction is largely attributable to left ventricular hypertrophy and contributes to hypotension during fluid removal on haemodialysis. Systolic dysfunction may be due to:
Left ventricular hypertrophy is a risk factor for early death in CKD, as in the general population. Systolic dysfunction is also a marker for early death.
Coronary artery calcification. Traditional risk factors (e.g. smoking, diabetes) can only partly explain the risk in patients with chronic nephropathies.
Coronary artery calcification is more common in patients with ESKD than in normal individuals and it is highly likely that this contributes significantly to cardiovascular mortality. Vascular calcification is frequent in all sizes of vessel in CKD.
In addition to the classical risk factors for atherosclerosis:
A raised (calcium × phosphate) product causes medial calcification.
Hyperparathyroidism may also contribute independently to the pathogenesis by increasing intracellular calcium.
Vascular calcification in uraemia is now thought to be an active process whereby vascular smooth muscle cells acquire osteoblast-like characteristics, possibly in response to elevated phosphate or (calcium × phosphate) product.
Inflammation is a potent mediator of vascular calcification by inhibition of fetuin (a glycoprotein synthesized by liver, which is a potent inhibitor of vascular calcification).
The impact of vascular calcification is the reduction of vascular compliance, which manifests by increased pulse pressure and pulse wave velocity, and increased afterload contributes further to left ventricular hypertrophy. In addition to myocardial abnormalities, vascular calcification with its associated biomechanical vessel wall alterations is a strong predictor of all-cause and cardiovascular morbidity and mortality in patients with CKD. Diffuse calcification of the myocardium is also common; the causes are similar.
Other cardiovascular risk factors. These include hyperhomocysteinaemia, Chlamydia pneumoniae infection, oxidative stress and elevated endogenous inhibitor of nitric oxide synthase and asymmetric dimethyl arginine (ADMA) levels. High ADMA levels in uraemia are in part caused by oxidative stress and can possibly explain the 52% increase in the risk of death and 34% increase in the risk of cardiovascular events in uraemic patients. The use of antioxidants, vitamin E or acetylcysteine has been associated with a significant reduction in all-cause and cardiovascular mortality. However, recent trials to reduce levels of homocysteine with folic acid, B6 and B12 supplementation have been unsuccessful.
The conclusion from a recent study of heart and renal protection (SHARP) in over 9500 CKD patients was that around one-quarter of all heart attacks, strokes, and revascularizations could be avoided in CKD by using a combination of ezetimibe and simvastatin to lower blood cholesterol. This combination however did not confer any survival advantage or prevent the development of ESKD.
Pericarditis
This is common and occurs in two clinical settings:
Uraemic pericarditis is a feature of severe, pre-terminal uraemia or of underdialysis. Haemorrhagic pericardial effusion and atrial arrhythmias are often associated. There is a danger of pericardial tamponade, and anticoagulants should be used with caution. Pericarditis usually resolves with intensive dialysis.
Dialysis pericarditis occurs as a result of an intercurrent illness or surgery in a patient receiving apparently adequate dialysis.
Management
Successful renal transplantation improves some, but not all, of the complications of CKD, therefore attempts should be made to prevent these complications by careful monitoring with ECG, echocardiography, angiography (if necessary) and nuclear imaging. CT (spiral CT) and/or MRI are useful in the assessment of arterial calcification. Treatment is with the control of risk factors (p. 608) as well as the treatment of hypercalcaemia and hyperparathyroidism (p. 624).
Progression of chronic kidney disease
CKD tends to progress inexorably to ESKD, although the rate of progression may depend upon the underlying nephropathy. Patients with chronic glomerular diseases tend to deteriorate more quickly than those with chronic tubulointerstitial nephropathies. Hypertension and heavy proteinuria are bad prognostic indicators. A nonspecific renal scarring process common to renal disorders of different aetiologies may be responsible for progression.
Possible causes of glomerular scarring and proteinuria include:
A rise in intraglomerular capillary pressure
Adaptive glomerular hypertrophy due to reduced arteriolar resistance and increased glomerular blood flow when there is reduced nephron mass.
This glomerular hyperfiltration, in response to nephron loss, was postulated as a common pathway for the progression of CKD.
Since the afferent arteriolar tone decreases more than efferent arteriolar tone, intraglomerular pressure and the amount of filtrate formed by a single nephron rises. Angiotensin II produced locally modulates intraglomerular capillary pressure and GFR, predominantly causing vasoconstriction of postglomerular arterioles, thereby increasing the glomerular hydraulic pressure and filtration fraction (Fig. 12.48). In addition, by its effect on mesangial cells and podocytes, it increases the pore sizes and impairs the size-selective function of basement membrane for macromolecules.
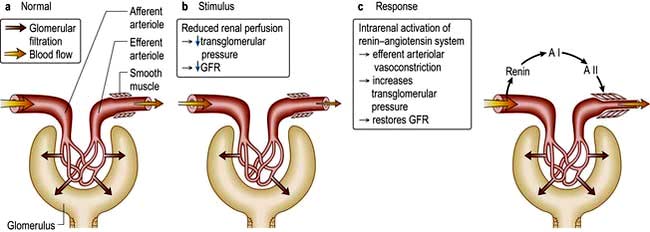
Figure 12.48 Glomerular dynamics: effect of the renin-angiotensin system. AI, angiotensin I; AII, angiotensin II.
Angiotensin II also modulates cell growth directly and indirectly by upregulating TGF-β, a potent fibrogenic cytokine, increases collagen synthesis and also causes epithelial cell transdifferentiation to myofibroblasts which contribute to excessive matrix formation. Furthermore, angiotensin II by upregulating plasminogen activator inhibitor-1 (PAI-1) inhibits matrix proteolysis by plasmin, resulting in accumulation of excessive matrix and scarring both in the glomeruli and interstitium.
Renal interstitial scarring is also a factor. The cause of this progressive interstitial damage and fibrosis is multifactorial. In addition to non-haemodynamic effects of angiotensin II, proteinuria per se (by exposing tubular cells to albumin and its bound fatty acids and cytokines) promotes secretion of pro-inflammatory mediators, which promote interstitial inflammatory cell infiltrate and further augment fibrosis and progression of CKD. The prognosis for renal function in chronic glomerular disorders is judged more accurately by interstitial histological appearances than by glomerular morphology.
Therapeutic manoeuvres (Box 12.6) aimed at inhibiting angiotensin II and reducing proteinuria mainly by ACEI and angiotensin-receptor antagonists have beneficial effects in slowing the rate of progression of CKD in both diabetic and non-diabetic renal diseases in humans.
 Box 12.6
Box 12.6
Renoprotection
Treatment
Patients with chronic kidney disease and proteinuria >1 g/24 h:
ACE inhibitor increasing to maximum dose
Add angiotensin receptor antagonist if goals are not achieveda
Add diuretic to prevent hyperkalaemia and help to control BP
Add calcium-channel blocker (verapamil or diltiazem) if goals not achieved
a In type 2 diabetes start with angiotensin receptor antagonist.
Management of chronic kidney disease
The underlying cause of CKD should be treated aggressively wherever possible.
Renoprotection
The multidrug approach to chronic nephropathies has been formalized in an international protocol (Box 12.6).
Correction of complications
Hyperkalaemia
Hyperkalaemia often responds to dietary restriction of potassium intake. Drugs which cause potassium retention (see p. 655) should be stopped. Occasionally, it may be necessary to prescribe ion-exchange resins to remove potassium in the gastrointestinal tract. Emergency treatment of severe hyperkalaemia is described on page 655.
Acidosis
Correction of acidosis helps to correct hyperkalaemia in CKD, and may also decrease muscle catabolism. Sodium bicarbonate supplements are often effective (4.8 g (57 mmol) of Na+ and HCO3– daily), and the possibility of oedema and hypertension owing to the so-called ‘extracellular fluid expansion’ was not borne out by a trial. Interestingly, correction of metabolic acidosis by sodium bicarbonate at a mean dose of 1.8 g/day was also associated with marked reduction in the rate of progression of CKD and development of ESKD in patients with stage 4 and 5 CKD. Calcium carbonate, also used as a calcium supplement and phosphate binder, has a beneficial effect on acidosis.
Calcium and phosphate control and suppression of PTH
Hypocalcaemia and hyperphosphataemia should be treated aggressively, preferably with regular (e.g. 3-monthly) measurements of serum PTH to assess how effectively hyperparathyroidism is being suppressed. Suppression of PTH levels to below two or three times the upper limit of ‘normal’ carries a high risk of development of a dynamic bone disease.
Dietary restriction of phosphate is seldom effective alone, because so many foods contain it. Oral calcium carbonate or acetate reduces absorption of dietary phosphate but is contraindicated where there is hypercalcaemia or hypercalciuria. Aluminium-containing gut phosphate binders are very effective but absorption of aluminium poses the risk of aluminium bone disease and development of cognitive impairment. They are now rarely used in the developed countries but are still used in the developing countries because they are extremely cheap.
Treatment
Gut phosphate binders. The polymer sevelamer reduces the calcium load and attenuates vascular calcification and also lowers cholesterol levels by 10%. However, it has not been shown to reduce mortality. Lanthanum carbonate is a non-calcium, non-aluminium phosphate binder that is effective and has a good safety profile.
Nicotinamide, an alternative to phosphate binders, blocks the intestinal sodium/inorganic phosphate (Na/Pi) cotransporter. It reduces phosphate levels and PTH levels alongside improvement in the lipid profile in dialysis patients.
Calcitriol (1,25-dihydroxycholecalciferol) or a vitamin D analogue, such as alfacalcidol, used in early CKD has no deleterious effect upon renal function provided hypercalcaemia is avoided. New vitamin D metabolites (22-oxacalcitriol, paricalcitol, doxercalciferol) are less calcaemic. However, with the exception of paricalcitol (19-nor-1,25 dihydroxyvitamin D2, which may have survival advantage), their usefulness over conventional but less expensive calcitriol or alfacalcidol remains to be established. Treatment with vitamin D analogues should be started only if serum PTH level is three times or more above the upper limit of normal, in order to prevent the development of adynamic bone disease (see p. 619). Vitamin D therapy has the disadvantage that it increases not only calcium but also phosphate absorption and may therefore exacerbate hyperphosphataemia and ectopic calcification including calciphylaxis (calcification of small vessels).
Calcimimetic agents (e.g. cinacalcet, a calcium-sensing receptor agonist, see p. 621) have also been tried in established secondary hyperparathyroidism with successful suppression of PTH levels and lowering of calcium × phosphate product. The long-term safety and efficacy of these agents has recently been confirmed and several observational studies have demonstrated that use of cinacalcet is associated with survival advantage.
Drug therapy
This should be minimized in patients with CKD. Tetracyclines (with the possible exception of doxycycline) should be avoided in view of their anti-anabolic effect and tendency to worsen uraemia. Drugs excreted by the kidneys, such as gentamicin, should be prescribed only in the absence of any alternative and drug levels monitored if feasible. Non-steroidal anti-inflammatory drugs (NSAIDs) should be avoided. Potassium-sparing agents, such as spironolactone and amiloride, pose particular dangers, as do artificial salt substitutes, all of which contain potassium.
Bardoxolone, an antioxidant inflammatory modulator, has shown a reduction in GFR in diabetic CKD.
Anaemia
The anaemia of erythropoietin (EPO) deficiency can be treated with synthetic (recombinant) human EPO (epoetin-α or -β, or the longer-acting darbepoetin-α or polyethylene glycol-bound epoetin-β). The intravenous route is used, initially with 50 U/kg of epoetin-α over 1–5 min, three times weekly. Subcutaneous administration of epoetin-α can also be used (see below). Blood pressure, haemoglobin concentration and reticulocyte count are measured every 2 weeks and the dose adjusted to maintain a target haemoglobin of 110–120 g/L. Darbepoetin-α can be used once a week. Continuous erythropoiesis receptor activator (CERA), a recently licensed epoetin in Europe, is a pegylated epoetin-β and can be given once a month. In trials, CERA has a similar activity to other types of epoetin. However, CERA use is extremely limited and data about loss of flexibility of dosing, overshooting the target haemoglobin and its efficacy in combination with short-acting epoetin are required before its widespread use.
The target haemoglobin to be achieved during the treatment of anaemia is being revised to between 110 and 120 g/L as studies in pre-dialysis CKD patients have not revealed any outcome benefits in patients who were treated to achieve higher haemoglobin targets (>120 g/L).
Failure to respond to 300 U/kg weekly, or a fall in haemoglobin after a satisfactory response, may be due to iron deficiency, bleeding, malignancy, infection, inflammation or formation of anti-EPO neutralizing antibodies. The demand for iron by the bone marrow is enormous when erythropoietin is commenced. Patients on EPO therapy are regularly monitored for iron status and considered iron deficient if plasma ferritin is <100 µg/L, hypochromic RBCs >10%, transferrin saturation <20%. Functional iron deficiency due to poor mobilization of iron, despite adequate iron stores (ferritin >500 µg/L) and a transferrin saturation of >20%, is a common finding in patients with chronic inflammation. It is caused by hepcidin (see p. 377), an acute phase reactant produced by the liver in response to cytokines, particularly IL-6. Intravenous (rather than oral) iron supplements optimize response to EPO treatment by repletion of iron stores. A recent randomized trial has demonstrated a beneficial effect of i.v. iron even in patients with ferritin >800 µg/L and transferrin saturation of 20%.
Correction of anaemia with EPO improves quality of life, exercise tolerance and sexual and cognitive function in dialysis patients, and leads to regression of left ventricular hypertrophy. Avoidance of blood transfusion also reduces the chance of sensitization to HLA antigens, which may otherwise be a barrier to successful renal transplantation.
The disadvantages of erythropoietin therapy are that it is expensive and causes a rise in blood pressure in up to 30% of patients, particularly in the first 6 months. Peripheral resistance rises in all patients, owing to loss of hypoxic vasodilatation and to increased blood viscosity. A rare complication is encephalopathy with fits, transient cortical blindness and hypertension. Several reports of anti-EPO antibody-mediated pure red-cell aplasia in patients receiving subcutaneous EPO therapy (particularly EPO-α) have been described. The exact cause is unknown but interventions such as using the intravenous route and changes in manufacture of prefilled syringes have reduced the number of cases by 80%. Other causes of anaemia should be looked for and treated appropriately (see p. 375).
Several erythropoiesis-stimulating agents are in clinical trials. An EPO mimetic is an engineered peptide which stimulates EPO receptor but still has to be given by injection. Oral agents which inhibit prolyl hydroxylase and prolong the life of HIF (hypoxia inducible factors) 1α, a transcription factor for endogenous production of EPO, have shown promise in phase 2 trials.
Male erectile dysfunction
Testosterone deficiency should be corrected. The oral phosphodiesterase inhibitors, e.g. sildenafil, tadalafil and vardenafil, are effective in ESKD and are the first-line therapy. The use of nitrates is a contraindication to this treatment. Other treatments are discussed on page 966.
Early referral of patients with chronic kidney disease
Patients need time to adjust to the demands of CKD and its treatment, and to absorb information. Veins required in the future for fashioning of an arteriovenous fistula should not be rendered useless by cannulation (Fig. 12.49).
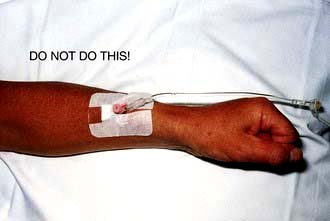
Figure 12.49 Intravenous cannula in exactly the WRONG place, in a right-handed patient with CKD who will in future need a left (non-dominant arm) radiocephalic fistula.
If the patient opts for regular haemodialysis, fashioning of an arteriovenous fistula should be carried out well in advance of the need for dialysis, when serum creatinine is of the order 400–500 µm in non-diabetics and at an even earlier stage in diabetics with poorer vasculature. Such fistulae require several weeks to mature and become usable for vascular access.
Renal replacement therapy
Approximately 100 white individuals per million population commence renal replacement therapy in the UK each year. The corresponding figure in black Africans and Asians in the UK is three to four times higher, largely owing to diabetic and hypertensive nephropathy. The aim of all renal replacement techniques is to mimic the excretory functions of the normal kidney, including excretion of nitrogenous wastes, maintenance of normal electrolyte concentrations, and maintenance of a normal extracellular volume.
Haemodialysis
Basic principles
In haemodialysis, blood from the patient is pumped through an array of semipermeable membranes (the dialyser, often called an ‘artificial kidney’), which bring the blood into close contact with dialysate, flowing countercurrent to the blood. The plasma biochemistry changes towards that of the dialysate owing to diffusion of molecules down their concentration gradients (Fig. 12.50).
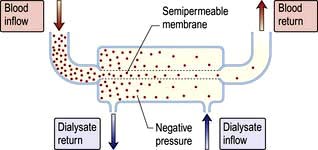
The dialysis machine comprises a series of blood pumps, with pressure monitors and bubble detectors and a proportionating unit, also with pressure monitors and blood leak detectors. Blood flow during dialysis is usually 200–300 mL/min and the dialysate flow usually 500 mL/min. The efficiency of dialysis in achieving biochemical change depends on blood and dialysate flow and the surface area of the dialysis membrane.
Dialysate is prepared by a proportionating unit, which mixes specially purified water with concentrate, resulting in fluid with the composition described in Table 12.24.
Table 12.24 Range of concentrations (mmol/L) in routinely available final dialysates used for haemodialysis
Sodium |
130–145 |
Potassium |
0.0–4.0 |
Calcium |
1.0–1.6 |
Magnesium |
0.25–0.85 |
Chloride |
99–108 |
Bicarbonate |
35–40 |
Glucose |
0–10 |
Access for haemodialysis
Adequate dialysis requires a blood flow of at least 200 mL/min. The most reliable long-term way of achieving this is surgical construction of an arteriovenous fistula (Fig. 12.51), using the radial or brachial artery and the cephalic vein. This results in distension of the vein and thickening (‘arterialization’) of its wall, so that after 6–8 weeks large-bore needles may be inserted to take blood to and from the dialysis machine. In patients with poor-quality veins or arterial disease (e.g. diabetes mellitus), arteriovenous polytetrafluoroethylene (PTFE) grafts are used for access. However, these grafts have a very high incidence of thrombosis and 2-year graft patency is only 50–60%. Dipyridamole or fish oils improve graft patency but warfarin, aspirin and clopidogrel do not and are associated with a high incidence of complications. In patients with PTFE stenosis, balloon angioplasty followed by stenting is associated with better long-term patency rates compared with angioplasty alone.
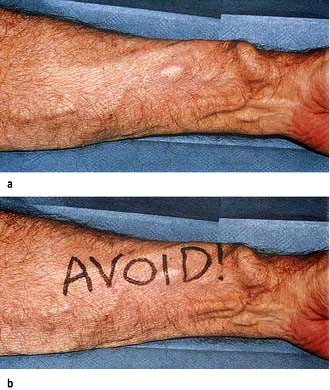
Figure 12.51 Arteriovenous (radiocephalic) fistula. (a) In the left forearm. (b) One way of reminding the anaesthetic, nursing and surgical team about the presence of an existing arteriovenous fistula in a haemodialysis patient about to undergo surgery. In particular, the fistula should not be compressed during surgery to avoid thrombosis within it.
If dialysis is needed immediately, a large-bore double lumen cannula may be inserted into a central vein – usually the subclavian, jugular or femoral. Semipermanent dual-lumen venous catheters can also be used, usually inserted via a skin tunnel to lessen the risk of infection. Stenosis of the subclavian vein is common, and the jugular route is preferred. However, they are associated with particularly high rates of infection, hospitalization, and death. These catheters are prone to partial or total occlusion, which may lead to inadequate dialysis and missed dialysis sessions and should be avoided whenever possible.
In a recent study a simple regimen in which recombinant tissue plasminogen activator (rt-PA) was used for locking the dialysis catheter once a week (with heparin used for the other two treatments each week) was superior to a regimen in which heparin locks were used after all the treatments; the rate of access failure was halved, and access-related infection was reduced by two-thirds.
Dialysis prescription
Dialysis must be tailored to an individual patient to obtain optimal results.
Initiation of dialysis
In clinical practice there is a wide variation in the timing of the starting dialysis therapy in patients with stage 5 CKD. There is a general tendency to start dialysis earlier with eGFR close to 15 mL/min rather then below 10 mL/min, despite a recent study showing that early initiation of dialysis was not associated with an improvement in survival or clinical outcomes. This underscores the fact that decisions about the timing of starting dialysis therapy should be taken individually rather than by a numerical parameter.
Dry weight
This is the weight at which a patient is neither fluid overloaded nor depleted. Patients are weighed at the start of each dialysis session and the transmembrane pressure adjusted to achieve fluid removal equal to the amount by which they exceed their dry weight.
The dialysate buffer
The dialysate buffer is usually acetate or bicarbonate. The sodium and calcium concentrations of the dialysate buffer are carefully monitored. A high dialysate sodium causes thirst and hypertension. A high dialysate calcium causes hypercalcaemia, while a low-calcium dialysate combined with poor compliance of medication with oral calcium carbonate and vitamin D may result in hyperparathyroidism.
Frequency and duration
Frequency and duration of dialysis are adjusted to achieve adequate removal of uraemic metabolites and to avoid excessive fluid overload between dialysis sessions. An adult of average size usually receives 4–5 hours’ treatment three times a week. Twice-weekly dialysis is adequate only if the patient has considerable residual renal function. However, in a recent study patients treated with haemodialysis six times a week benefited more with respect to the composite outcomes of death or change in left ventricular mass and quality of life but required more frequent interventions related to vascular access.
Short-duration dialysis using very biocompatible high-flux membranes is commonly employed. Advantages include shorter duration of treatment and hence increased patient convenience. Disadvantages include higher cost of the membranes employed and, in all probability, higher prevalence of hypertension in such patients, requiring hypotensive medication. It should not be forgotten that normal kidneys work for 24 hours a day, 7 days a week, and that dialysis is a poor substitute for the natural state.
Adequate/optimal dialysis should be adjusted to individual patients’ needs. All patients are anticoagulated (usually with heparin) during treatment as contact with foreign surfaces activates the clotting cascade. In the UK, a small number of patients manage self-supervised home haemodialysis.
Complications
Hypotension during dialysis is the major complication. Contributing factors include: an excessive removal of extracellular fluid, inadequate ‘refilling’ of the blood compartment from the interstitial compartment during fluid removal, abnormalities of venous tone, autonomic neuropathy, acetate intolerance (acetate acts as a vasodilator) and left ventricular hypertrophy.
Very rarely, patients may develop anaphylactic reactions to ethylene oxide, which is used to sterilize most dialysers. Patients receiving ACE inhibitors are at risk of anaphylaxis if polyacrylonitrile dialysers are used.
Other potential, rare, complications include the hard-water syndrome (caused by failure to soften water resulting in a high calcium concentration prior to mixing with dialysate concentrate), haemolytic reactions and air embolism.
Adequacy of dialysis
Dialysis treatment is empirical since the size, number and nature of ‘uraemic toxins’ is unclear. The only true measure of adequacy is patient mortality and morbidity. Adequate nutrition of the patient, as well as adequate dialysis is necessary to reduce morbidity and mortality.
Symptoms of underdialysis are nonspecific and include insomnia, itching and fatigue, despite adequate correction of anaemia, restless legs and a peripheral sensory neuropathy.
Adequacy of dialysis may be assessed by computerized calculation of urea kinetics, requiring measurement of the residual renal urea clearance, the rate of rise of urea concentration between dialysis sessions, and the reduction in urea concentration during dialysis. The dialysis dose is normally defined in terms of urea reduction ratio (URR) and/or equilibrated urea clearance, eKt/V (where K is the dialyser clearance, t is the duration of dialysis in minutes, and V is the urea distribution volume estimated as total body water). Kt/V of 1.0–1.2 and/or URR of 65% per dialysis session is the minimum threshold required for well-nourished dialysis patients dialysed three times per week. It is unclear whether a higher eKt/V is associated with a better outcome, although no additional benefits of high (1.53) compared with standard (1.16) eKt/V were seen over 5 years’ follow-up. It is likely that duration of haemodialysis session is a factor in itself in addition to the efficiency with which small molecules such as urea are cleared. The best haemodialysis outcome data are seen in renal units where long hours (8 h per session) of dialysis are routinely practised.
Haemodialysis is the most efficient way of achieving rapid biochemical improvement, for instance in the treatment of acute kidney injury or severe hyperkalaemia. This advantage is offset by disadvantages such as haemodynamic instability, especially in acutely ill patients with multiorgan disease, and over-rapid correction of uraemia can lead to ‘dialysis disequilibrium’. This is characterized by nausea and vomiting, restlessness, headache, hypertension, myoclonic jerking, and in severe instances seizures and coma owing to rapid changes in plasma osmolality leading to cerebral oedema.
These problems have led to the increasing adoption of gentler continuous methods for the treatment of acute kidney injury (see below).
Haemofiltration
This involves removal of plasma water and its dissolved constituents (e.g. K+, Na+, urea, phosphate) by convective flow across a high-flux semipermeable membrane, and replacing it with a solution of the desired biochemical composition (Fig. 12.52). Lactate is used as buffer in the replacement solution because rapid infusion of acetate causes vasodilatation, and bicarbonate may cause precipitation of calcium carbonate.
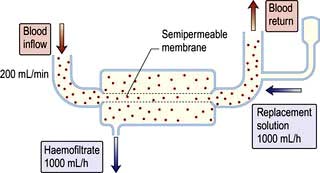
Haemofiltration can be used for both acute and CKD and is used in mainland Europe for CKD patients with haemodynamic instability. High volumes need to be exchanged in order to achieve adequate small molecule removal, typically a 22 L exchange three times a week for maintenance treatment and 1 L per hour in acute kidney injury. Financial costs of disposable items (such as filters and replacement fluid) are high and only a selected group of patients with ESKD are managed in this way. Modern dialysis machines have built-in facilities to generate online ultrapure water, which has minimized the cost of the procedure and given an option to the clinician to use this technique either as haemofiltration or in combination with dialysis as haemodiafiltration to increase middle molecule clearance (e.g. β2-microglobulin) and prevent long-term dialysis complications such as dialysis-related amyloidosis, particularly in young, highly sensitized, non-transplantable patients.
Peritoneal dialysis
Peritoneal dialysis utilizes the peritoneal membrane as a semipermeable membrane, avoiding the need for extracorporeal circulation of blood. This is a very simple, low-technology treatment compared to haemodialysis. The principles are simple (Fig. 12.53).
A tube is placed into the peritoneal cavity through the anterior abdominal wall (Fig. 12.54).
Dialysate is run into the peritoneal cavity, usually under gravity.
Urea, creatinine, phosphate and other uraemic toxins pass into the dialysate down their concentration gradients.
Water (with solutes) is attracted into the peritoneal cavity by osmosis, depending on the osmolarity of the dialysate. This is determined by the glucose or polymer (icodextrin) content of the dialysate (Table 12.25).
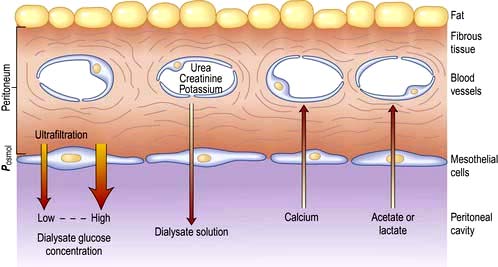
Figure 12.53 Principles of peritoneal dialysis. Water is attracted into the peritoneal cavity depending on the osmolarity of the dialysate.
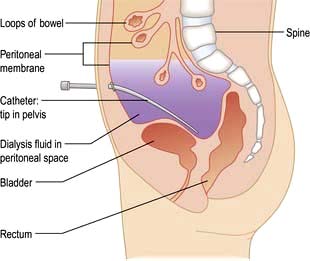
Table 12.25 Range of concentrations (mmol/L) in routinely available CAPD dialysate
Sodium |
130–134 |
Potassium |
0 |
Calcium |
1.0–1.75 |
Magnesium |
0.25–0.75 |
Chloride |
95–104 |
Lactate |
35–40 |
Glucosea |
77–236 |
Total osmolality |
356–511 mOsm/kg |
a Glucose content is often expressed as g/dL of anhydrous glucose (e.g. 1.36% = 77 mmol/L). An even more hypertonic dialysate (6.36%) is available for acute (intermittent) peritoneal dialysis.
Chronic peritoneal dialysis requires insertion of a soft catheter, with its tip in the pelvis, exiting the peritoneal cavity in the midline and lying in a skin tunnel with an exit site in the lateral abdominal wall (Fig. 12.54).
This form of dialysis can be adapted in several ways.
Continuous ambulatory peritoneal dialysis (CAPD). Dialysate is present within the peritoneal cavity continuously, except when dialysate is being exchanged. Dialysate exchanges are performed three to five times a day, using a sterile no-touch technique to connect 1.5–3 L bags of dialysate to the peritoneal catheter; each exchange takes 20–40 min. This is the technique most often used for maintenance peritoneal dialysis in patients with ESKD.
Nightly intermittent peritoneal dialysis (NIPD), also called automated peritoneal dialysis (APD). An automated device is used to perform exchanges each night while the patient is asleep. Sometimes dialysate is left in the peritoneal cavity during the day in addition, to increase the time during which biochemical exchange is occurring. Few trials have demonstrated superiority of APD over CAPD with regard to complications such as peritonitis, fluid status and in anuric patients.
Tidal dialysis. A residual volume is left within the peritoneal cavity with continuous cycling of smaller volumes in and out.
Osmotic removal of excess plasma water and solutes is achieved using hypertonic dialysate (either due to high glucose concentration or icodextrin), which exerts an osmotic ‘drag’. Depending on the patient’s fluid intake and residual urine output, it may be necessary to use one or more hypertonic dialysate bags daily to achieve fluid balance in CAPD. Fluid overload is a relatively common problem in CAPD, and is due to failure of transport across the peritoneal membrane.
Complications
Peritonitis
Bacterial peritonitis is the most common serious complication of CAPD and other forms of peritoneal dialysis. Clinical presentations include abdominal pain of varying severity (guarding and rebound tenderness are unusual), and a cloudy peritoneal effluent, without which the diagnosis cannot be made. Microscopy reveals a neutrophil count above 100 cells/mL. Nausea, vomiting, fever and paralytic ileus may be seen if peritonitis is severe. The incidence of CAPD-associated peritonitis has been much reduced (to about one episode every 2 patient-years) by use of a Y-disconnect system in preference to previous methods.
CAPD peritonitis must be investigated with culture of peritoneal effluent. Empirical antibiotic treatment is started, with a spectrum which covers both Gram-negative and Gram-positive organisms. Antibiotics may be given by the oral, intravenous or intraperitoneal route; most centres rely on intraperitoneal antibiotics. Common causative organisms are listed in Table 12.26.
Table 12.26 Some causes of CAPD peritonitisa
| Approximate cases (%) | |
|---|---|
Staphylococcus epidermidis |
40–50 |
Escherichia coli, Pseudomonas and other Gram-negative organisms |
25 |
Staphylococcus aureus |
15 |
Mycobacterium tuberculosis |
2 |
Candida and other fungal species |
2 |
a In approximately 20%, no bacteria are found.
Staph. aureus peritonitis should lead to a search for nasal carriage of this organism, and Staph. epidermidis peritonitis may indicate contamination from the patient’s (or helper’s) skin. Relapsing Staph. epidermidis peritonitis with an organism with the same antibiotic sensitivity pattern on each occasion may indicate that the Tenckhoff catheter has become colonized: often this is difficult to eradicate without replacement of the catheter under antibiotic cover.
Gram-negative peritonitis may complicate septicaemia from urinary or bowel infection. A mixed growth of Gram-negative and anaerobic organisms strongly suggests bowel perforation, and is an indication for laparotomy.
Fungal peritonitis often follows antibacterial treatment but may occur de novo. Clinical presentation is very variable. It is rare to be able to cure fungal peritonitis without catheter removal as well as antifungal treatment. Intraperitoneal amphotericin has been associated with the formation of peritoneal adhesions.
Infection around the catheter site
Infection where the catheter exits through the skin is relatively common. It should be treated aggressively (with systemic and/or local antibiotics) to prevent spread of the infection into the subcutaneous tunnel and the peritoneum. The most common causative organisms are staphylococci, including MRSA. Exit site infections can be reduced by routine use of mupirocin or gentamicin ointments locally and intranasally in those with colonized nares. Exit site infection due to Pseudomonas is treated by antibiotics and resiting the exit following catheter exchange.
Other complications
CAPD is often associated with constipation, which in turn may impair flow of dialysate in and out of the pelvis. Occasionally, dialysate may leak through a diaphragmatic defect into the thoracic cavity, causing a massive pleural ‘effusion’. The glucose content of the effusion is usually diagnostic, or the diagnosis may be made by instillation of methylthioninium chloride (methylene blue) with dialysate and the demonstration of a blue colour on pleural tap. Dialysate may also leak into the scrotum down a patent processus vaginalis.
Failure of peritoneal membrane function is a predictable complication of long-term CAPD, resulting in worsening biochemical exchange and decreased ultrafiltration with hypertonic dialysate. It is thought that this problem may be accelerated by excessive reliance on hypertonic dialysate to remove fluid.
Sclerosing peritonitis is a potentially fatal complication of CAPD. The cause is often unclear, but recurrent peritonitis and exposure of the peritoneum to unphysiological high glucose concentrations is responsible in most cases. Progressive thickening of the peritoneal membrane occurs in association with adhesions and strictures, turning the small bowel into a mass of matted loops and causing repeated episodes of small bowel obstruction. CAPD should be abandoned. Improvement may follow renal transplantation or treatment with prednisolone or azathioprine.
Contraindications
There are few absolute contraindications apart from unwillingness or inability on the patient’s part to learn the technique.
Previous peritonitis causing peritoneal adhesions may make peritoneal dialysis impossible, but the extent of adhesions is difficult to predict and it may be worth an attempted surgical placement of a dialysis catheter.
The presence of a stoma (colostomy, ileostomy, ileal urinary conduit) makes successful placement of a dialysis catheter extremely unlikely.
Active intra-abdominal sepsis, for instance due to diverticular abscesses, is an absolute contraindication to peritoneal dialysis although diverticular disease per se is not.
Abdominal hernias may often expand during CAPD as a result of increased intra-abdominal pressure, and should ideally be repaired before or at the time of CAPD catheter insertion.
Visual impairment may make it difficult for a patient to perform dialysate exchanges, but completely blind patients can be trained in the technique if adequately motivated.
Severe arthritis makes it difficult to perform the exchanges, but a large number of mechanical aids are available. Sterilization of connections by heat or ultraviolet light reduces the risk of peritonitis.
Some studies suggest that ESKD patients with coronary artery disease or congestive heart failure have significantly higher morbidity and mortality on CAPD compared with HD. Moreover, elderly patients do worse on CAPD than younger patients. It is advisable to consider HD as the first-choice dialysis modality in these patients where it is possible.
Adequacy of peritoneal dialysis
No consensus exists on the optimum degree of removal of urea and other waste products to be obtained in unit time but there is general agreement that minimum dose delivered should not be <1.7. Based on observational studies, weekly Kt/V of 2.0 coupled with a creatinine clearance target of 60 L per week had been recommended. However, in a randomized prospective study (ADEMEX), standard daily exchanges of 8 L in total (creatinine clearance 40 L per week) compared with a creatinine clearance of 60 L per week yielded similar patient and technique survival. Peritoneal dialysis inadequacy becomes common when residual renal function declines to zero. With increasing time on treatment, adequacy may become impaired owing to alterations in the efficiency of the peritoneal membrane in transporting waste products, fluid and electrolytes.
Complications of all long-term dialysis
Cardiovascular disease (see p. 620) and sepsis are the leading causes of death in long-term dialysis patients.
Causes of fatal sepsis include peritonitis complicating peritoneal dialysis and Staph. aureus infection (including endocarditis) complicating the use of indwelling access devices for haemodialysis.
Dialysis amyloidosis
This is the accumulation of amyloid protein (p. 1043) as a result of failure of clearance of β2-microglobulin, a molecule of 11.8 kDa. This protein is the light chain of the class I HLA antigens and is normally freely filtered at the glomerulus but is not removed by cellulose-based haemodialysis membranes. The protein polymerizes, possibly after modification by non-enzymic glycosylation, to form amyloid deposits, which may cause median nerve compression in the carpal tunnel or a dialysis arthropathy – a clinical syndrome of pain and disabling stiffness in the shoulders, hips, hands, wrists and knees. β2-microglobulin-related amyloid may be demonstrated in the synovium. Amyloid deposits (see p. 1043) can also cause pathological bone cysts and fractures, pseudotumours and gastrointestinal bleeding caused by amyloid deposition around submucosal blood vessels. The extent of amyloid deposition is best assessed by nuclear imaging, either using [99mTc] DMSA, or more specifically, by the use of radiolabelled serum amyloid P component.
Rapid improvement after renal transplantation is probably due to steroid therapy as low-dose prednisolone alone can also cause an improvement. A change to a biocompatible synthetic membrane has also been reported to be of benefit: again, the mechanism for this improvement is not clear. β2-microglobulin clearance is several times higher in patients treated with haemofiltration or haemodiafiltration and these techniques are increasingly used in patients at high risk of developing this complication.
Dialysis in the elderly
Dialysis can prolong life, but the benefit to the individual and particularly elderly patients, varies widely. Outcomes in frail elderly people who are undergoing dialysis are poor. Small studies suggest that mortality or quality-of-life outcomes do not differ significantly among selected patients who elect to undergo dialysis compared to those who decide against dialysis. In one study, over 50% died within the first year of initiating dialysis and around 30% had a decrease in functional status.
Prior to the initiation of dialysis, elderly patients must be informed about its modest benefit in their age group and the possibility of conservative therapy that does not involve dialysis. Conservative therapy should be discussed, not as a last resort but as a clear option that might be more effective in promoting patient goals. There is a need for well structured palliative care infrastructure staffed by trained individuals to deal with end of life decision issues in dialysis patients.
Transplantation
Successful renal transplantation offers the potential for almost complete rehabilitation in ESKD. This mode of renal replacement therapy has significant survival advantage compared to dialysis patients on transplant waiting lists. It allows freedom from dietary and fluid restriction; anaemia and infertility are corrected; and the need for parathyroidectomy is reduced. It is the treatment of choice for most patients with ESKD. The supply of donor organs (in the UK, 30/million per year) is greatly exceeded by demand (48/million per year), and donor organs are therefore scarce and a valuable resource that must be used optimally.
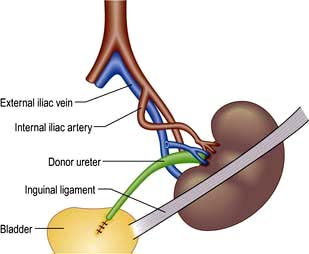
The technique involves the anastomosis of an explanted human kidney, usually either from a cadaveric donor or from a living close relative, on to the iliac vessels of the recipient (Fig. 12.55). The donor ureter is placed into the recipient’s bladder. Unless the donor is genetically identical (i.e. an identical twin), immunosuppressive treatment is needed, for as long as the transplant remains in place, to prevent rejection. Refinements in patient selection and assessment of donor–recipient compatibility, improvements in surgical techniques and the development of more efficient immunosuppressive regimens have increased patient and graft survival. Some 80% of grafts now survive for 5–10 years in the best centres, and 50% for 10–30 years. However, the half-life of renal allografts is still 13–16 years. The three most common causes of late graft loss are death with functioning graft, recurrence of renal disease and chronic allograft nephropathy.
Factors affecting success
ABO (blood group) compatibility between donor and recipient is required. However, in some centres, particularly in Japan where a cadaveric organ donation programme is not well established, ABO-incompatible renal transplants are increasingly performed. These transplants follow with immunoadsorption to remove preformed antibodies, splenectomy, anti-CD20 antibodies to remove B lymphocytes, and intravenous pooled immunoglobulins for immunomodulation or anti-idiotypic antibodies. In experienced hands, results are acceptable.
Matching donor and recipient for HLA type
Matching for HLA-DR antigens appears to have the most impact on graft survival. Studies have shown that matching at the HLA-B locus has only a minor effect on graft outcomes. Complete compatibility at A, B and DR offers the best chance of success, followed by a single HLA mismatch (i.e. antigen possessed by the donor and not possessed by the recipient). The effect of further degrees of mismatching upon graft survival in first transplants is of modest degree. Nationwide matching schemes for kidneys retrieved from cadaver donors are in existence. However, with availability of more efficient immunosuppressive agents, the value of HLA matching in the overall transplant outcome is, if any, only modest. Transplantation with completely mismatched kidneys, particularly when the donor is the patient’s partner, is routinely practised and results are as good as, if not better than, properly matched cadaveric kidneys.
A donor factor, an allotype of the C3 complement molecule, may be associated with better long-term outcomes for cadaveric kidney grafts. This report raised the possibility that the presence of the C3F (fast) allele in a kidney allograft is associated with a better outcome. However, this finding could not be reproduced in a subsequent study, which underscores the necessity of validation of unusual findings by several groups before adopting them in the clinical practice.
FURTHER READING
Varagunam M, Yaqoob MM, Döhler B et al. C3 polymorphisms and allograft outcome in renal transplantation. N Engl J Med 2009; 360(9):874–880.
Jardine AG et al. Prevention of cardiovascular disease in adult recipients of kidney transplants. Lancet 2011; 378:1419–1427.
Nankivell BJ, Kuypers DRJ. Diagnosis and prevention of chronic kidney allograft loss. Lancet 2011; 378:1428–1437.
The donor kidney
Cadaveric donation
Most countries allow the removal of kidneys and other organs from patients who have suffered irretrievable brain damage (‘brainstem death’) while their hearts are still beating (see p. 897). Due to shortage of solid organs and increasing numbers of patients waiting for them, several countries now allow the retrieval of organs after cardiac death, with comparable results to heart beating donations. Moreover, elderly donors (age >60 years) or those between the ages of 55 and 59 years but co-morbidity such as hypertension, diabetes, preretrieval AKI and intracranial haemorrhage as a cause of death known as expanded criteria donors (ECD) are increasingly used.
Living related donation
A close relative may volunteer as a potential donor. A sibling donor may be HLA identical or share one or no haplotypes with the potential recipient. In the UK, donor age must be 18 years or more.
Potential living related donors are subjected to an intensive preoperative evaluation, including clinical examination and measurement of renal function, tests for carriage of hepatitis B, C, HIV and cytomegalovirus, and detailed imaging of renal anatomy, to be sure that transplantation will be technically feasible.
Unrelated living donors may be accepted provided no inducement (financial or otherwise) is involved. Paid live non-related donor transplantation is illegal in the UK.
Immunosuppression for transplantation
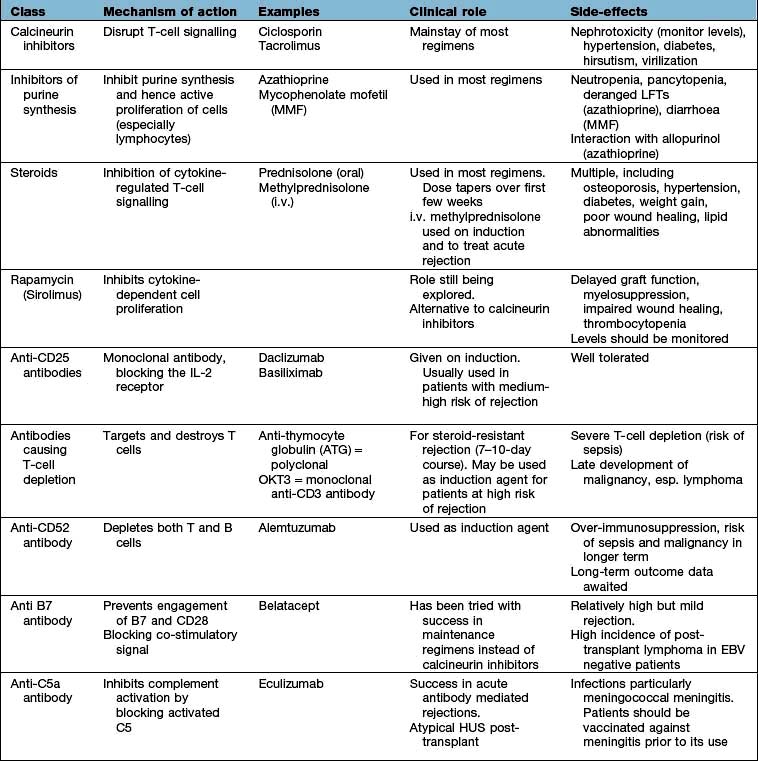
Long-term drug treatment for the prevention of rejection is employed in all cases apart from living related donation from an identical twin. Some degree of immunological tolerance does develop, and the risk of rejection is highest in the first 3 months after transplantation. In the early months, rejection episodes occur in less than 30% of cadaver kidney recipients. Most are reversible. A combination of immunosuppressive drugs is usually used (Table 12.27).
FURTHER READING
Ekberg H, Tedesco-Silva H, Demirbas A et al. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med 2007; 347:2562–2575.
Vincenti F, Charpentier B, Vanrenterghem Y et al. A phase III study of belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study). Am J Transplant 2010; 10:536–546.
Complications
Acute tubular necrosis (ATN)
ATN is the commonest cause of cadaveric graft dysfunction (up to 40–50%), particularly in kidneys from DCD or ECD donors and prolonged cold ischaemia time (>24 hours). Delayed graft function due to ATN is associated with worse long-term outcome and also predisposes the graft to rejection. It is usual practice to use induction with antibodies and use 50% of the starting dose of calcineurin inhibitors in recipient of grafts at high risk for ATN.
Technical failures
There may be occlusion or stenosis of the arterial anastomosis, occlusion of the venous anastomosis, and urinary leaks owing to damage to the lower ureter, or defects in the anastomosis between ureter and recipient bladder. If urine output drops, Doppler ultrasonography, DTPA scanning and/or renal angiography is used. Surgical reimplantation may be required.
Acute rejection (AR)
AR is seen in between 10% and 30% of transplant recipients and usually presents with declining renal function within the first 3 months. Renal biopsy confirms the diagnosis and also assesses the severity and type of rejection (cellular or vascular or antibody mediated, also called humoral rejection). Therapy in cellular rejection is high-dose pulse steroid; in acute vascular rejection (diagnosed by the presence of additional endothelial inflammation), ATG, anti-lymphocyte globulin (ALG) or OKT3 is used.
Histological appearances similar to vascular rejection are seen in a small number of patients where the culprit is an agonistic antibody directed against angiotensin receptor 1 (ATR 1), which usually presents with sudden decline in renal function but unlike vascular rejection also manifests with malignant phase hypertension and seizures. It responds to angiotensin receptor blockers in addition to conventional antirejection strategies (Fig. 12.56a). Humoral rejection (diagnosed by the presence of circulating donor-specific anti-HLA antibodies and evidence of complement activation on renal biopsy by C4 staining) (Fig. 12.56b) is usually treated empirically by a combination of i.v. polyclonal immunoglobulin infusion to neutralize and promote the clearance of anti-HLA antibodies, plasmapheresis (to remove antibodies) and anti-CD20 antibody administration (to deplete B lymphocytes) with variable success.
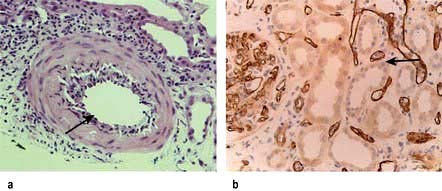
Figure 12.56 Histology of kidney in rejection. (a) Acute vascular rejection: the mononuclear inflammatory cell infiltrate is limited to the expanded intima and does not involve the entire vascular wall as in systemic vasculitis. (b) Antibody mediated rejection: an acute tubular necrosis with capillary glomerulitis and peritubular and glomerular capillary C4d positivity.
More than one rejection within the first 3 months, vascular and/or humoral rejection, delayed rejection (requirement of dialysis within the first week after transplantation), and failure of serum creatinine to return to baseline (<130 µmol/L) are associated with worse long-term outcome. The outcome of renal transplantation during an episode of acute rejection is difficult to predict, even with an allograft biopsy. Urinary levels of messenger RNA (mRNA) for FOXP3, a specification and functional factor for regulatory T lymphocytes, improve the prediction of outcome of acute rejection of renal transplants by a non-invasive means. It predicts with a reasonable degree of sensitivity and specificity successful outcome or otherwise in a renal transplant with acute rejections.
Infections
In the first month post-transplantation, infections tend to be from bacterial sources seen typically in the surgical population. Cytomegalovirus (CMV) infections develop weeks or months after transplantation in 70% of CMV-seronegative recipients receiving grafts from a seropositive donor and in patients receiving biological agents (antibodies) as induction or therapy for rejection, unless prophylaxis with valganciclovir is given. Prophylaxis is also routinely given against Pneumocystis jiroveci (co-trimoxazole) and oral candidiasis (nystatin or amphotericin lozenges). Polyomavirus infections (BK nephropathy) result in graft dysfunction and eventual loss due to mainly tubulointerstitial nephritis diagnosed by renal biopsy and presence of cellular inclusion bodies positive for SV40 (Fig. 12.57). There is no known specific treatment with the exception of tapering immunosuppression and with cautious use of leflunomide and cidofovir.
Post-transplantation lymphoproliferative disorders
Epstein–Barr virus-associated malignancies are common in patients who received biological agents and in children. Tapering of ciclosporin or tacrolimus, cessation of antiproliferative agents (azathioprine or MMF) and monitoring for the reappearance of cytotoxic lymphocytes has improved the outcome.
Chronic allograft nephropathy (CAN)
CAN remains the most common cause of late graft failure. The process is mediated by immunological possibly due to chronic antibody mediated rejection due to de novo synthesis of donor specific antibodies and non-immunological factors and results in a progressive irreversible decline in graft function with mild to modest proteinuria (<3 g/day). Unfortunately there is no established therapy of proven efficiency.
Malignancy
Immunosuppressive therapy increases the risk of skin tumours, including basal and squamous cell carcinoma. In white recipients, exposure to ultraviolet light should be minimized and sun-block creams employed. Other common cancers are renal, cervical and vaginal. In female recipients, regular yearly cervical smears should be carried out.
Cardiovascular disease
Cardiovascular disease is the cause of death post transplantation in 50% of cases. This is due to increased incidence of hypertension, obesity, diabetes and insulin resistance lipid disorders. Use of a statin (fluvastatin) was associated with reduction in cardiac endpoints (myocardial infarction or revascularization) post transplantation but overall mortality remained unchanged in a randomized prospective study.
Post-transplant osteoporosis
This is common following transplantation owing to treatment with steroids. Maximum bone loss occurs within the first 3 months and regular DXA scans are necessary. Bisphosphonates (alendronate, pamidronate) and alfacalcidol with or without calcium carbonate have proven to be effective in control studies.
Renal transplantation in HIV patients
Modern highly active antiretroviral therapy (HAART) offers patients with HIV infection a near-normal life expectancy. However, an increasing number develop end-stage CKD. Until recently, HIV was considered to be a contraindication to renal transplantation. Nevertheless, a recent study of outcomes for 150 HIV-infected patients undergoing renal transplantation has shown that kidney transplantation is safe and effective in patients, at least in the short term. The patients in the study had CD4+ T-cell counts ≥200/mL3 and undetectable plasma levels of HIV type 1 RNA while on stable HAART during the 16 weeks before renal transplantation. Median follow-up was 1.7 years. Survival rates at 1 year (95%) and 3 years (88%) were worse in HIV-infected patients than in the general population of kidney transplant recipients, but better than those in patients aged ≥65 years. Many rejection episodes were glucocorticoid-resistant, suggesting an aggressive response to donor antigens.