29 The gastrointestinal tract
Overview
In addition to its main function of digestion and absorption of food, the gastrointestinal tract is one of the major endocrine systems in the body and has its own integrative neuronal network, the enteric nervous system (see Ch. 12), which contains almost the same number of neurons as the spinal cord. It is also the site of many common pathologies, ranging from simple dyspepsia to complex autoimmune conditions such as Crohn’s disease. Medicines for treating these gastrointestinal disorders comprise some 8% of all prescriptions. In this chapter, we briefly review the physiological control of gastrointestinal function and then discuss the pharmacological characteristics of drugs affecting gastric secretion and motility, and those used to treat intestinal inflammation.
The Innervation and Hormones of the Gastrointestinal Tract
The blood vessels and the glands (exocrine, endocrine and paracrine) that comprise the gastrointestinal tract are under both neuronal and hormonal control.
Neuronal Control
There are two principal intramural plexuses in the tract: the myenteric plexus (Auerbach’s plexus) between the outer, longitudinal and the middle, circular muscle layers, and the submucous plexus (Meissner’s plexus) on the lumenal side of the circular muscle layer. These plexuses are interconnected, and their ganglion cells receive preganglionic parasympathetic fibres from the vagus, which are mostly cholinergic and excitatory, although a few are inhibitory. Incoming sympathetic fibres are largely postganglionic. In addition to innervating blood vessels, smooth muscle and some glandular cells directly, some sympathetic fibres may terminate in these plexuses, where they inhibit acetylcholine secretion (see Ch. 12).
The neurons within the plexuses constitute the enteric nervous system and secrete not only acetylcholine and noradrenaline (norepinephrine), but also 5-hydroxytryptamine, purines, nitric oxide and a variety of pharmacologically active peptides (see Chs 12–14, 16, 19 and 20). The enteric plexus also contains sensory neurons, which respond to mechanical and chemical stimuli.
Hormonal Control
The hormones of the gastrointestinal tract include both endocrine and paracrine secretions. The endocrine secretions (i.e. substances released into the bloodstream) are mainly peptides synthesised by endocrine cells in the mucosa. Important examples include gastrin and cholecystokinin. The paracrine secretions include many regulatory peptides released from special cells found throughout the wall of the tract. These hormones act on nearby cells, and in the stomach the most important of these is histamine. Some of these paracrine factors also function as neurotransmitters.
Orally administered drugs are, of course, absorbed in the gastrointestinal tract (Ch. 8). Other functions of the gastrointestinal tract that are important from the viewpoint of pharmacological intervention are:
Gastric Secretion
The stomach secretes about 2.5 litres of gastric juice daily. The principal exocrine components are proenzymes such as prorennin and pepsinogen elaborated by the chief or peptic cells, and hydrochloric acid (HCl) and intrinsic factor (see Ch. 25) secreted by the parietal or oxyntic cells. The production of acid is important for promoting proteolytic digestion of foodstuffs, iron absorption and killing pathogens. Mucus-secreting cells also abound among the surface cells of the gastric mucosa. Bicarbonate ions are secreted and trapped in the mucus, creating a gel-like protective barrier that maintains the mucosal surface at a pH of 6–7 in the face of a much more acidic environment (pH 1–2) in the lumen. Alcohol and bile can disrupt this layer. Locally produced ‘cytoprotective’ prostaglandins stimulate the secretion of both mucus and bicarbonate.
Disturbances in these secretory and protective mechanisms are thought to be involved in the pathogenesis of peptic ulcer, and indeed in other types of gastric damage such as gastro-oesophageal reflux disease (GORD)1 and injury caused by non-steroidal anti-inflammatory drugs (NSAIDs).
The Regulation of Acid Secretion by Parietal Cells
The regulation of acid secretion is important in the pathogenesis of peptic ulcer, and constitutes a particular target for drug action. The secretion of the parietal cells is an isotonic solution of HCl (150 mmol/l) with a pH less than 1, the concentration of hydrogen ions being more than a million times higher than that of the plasma. The Cl− is actively transported into canaliculi in the cells that communicate with the lumen of the gastric glands and thus with the stomach itself. This Cl− secretion is accompanied by K+, which is then exchanged for H+ from within the cell by a K+-H+-ATPase (the ‘proton pump’, Fig. 29.1). Carbonic anhydrase catalyses the combination of carbon dioxide and water to give carbonic acid, which dissociates into H+ and bicarbonate ions. The latter exchanges across the basal membrane of the parietal cell for Cl−. The principal mediators that directly—or indirectly—control parietal cell acid output are:
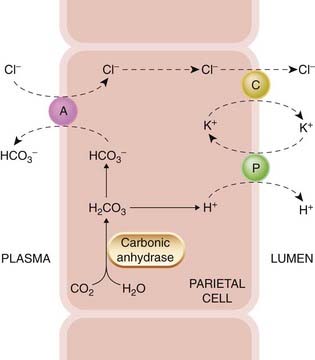
Fig. 29.1 A schematic illustration of the secretion of hydrochloric acid by the gastric parietal cell.
Secretion involves a proton pump (P), which is an H+-K+-ATPase, a symport carrier (C) for K+ and Cl−, and an antiport (A), which exchanges Cl− and HCO3−. An additional Na+/H+ antiport situated at the interface with the plasma may also have a role (not shown).
Histamine
Histamine is discussed in Chapter 26, and only those aspects of its pharmacology relevant to gastric secretion will be dealt with here. Neuroendocrine cells abound in the stomach and the dominant type are the ECL cells (enterochromaffin-like cells; histamine-containing cells similar to mast cells) which lie close to the parietal cells. They sustain a steady basal release of histamine, which is further increased by gastrin and acetylcholine. Histamine acts in a paracrine fashion on parietal cell H1 receptors, increasing intracellular cAMP. These cells are responsive to histamine concentrations that are below the threshold required for vascular H2 receptor activation.
Gastrin
Gastrin is synthesised by G cells in the gastric antrum and secreted into the portal blood (i.e. it acts in an endocrine fashion). Its main action is stimulation of acid secretion by ECL cells through its action at gastrin/cholecystokinin (CCK)2 receptors, which elevate intracellular Ca2+. Gastrin receptors also occur on the parietal cells but their significance in the control of physiological secretion is controversial. CCK2 receptors are blocked by the experimental drug proglumide (Fig. 29.2), which weakly inhibits gastrin action.
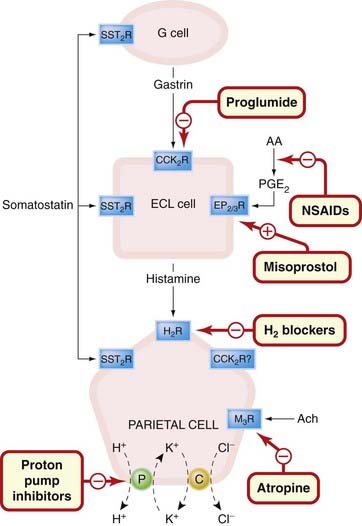
Fig. 29.2 Schematic diagram showing the regulation of the acid-secreting gastric parietal cell, illustrating the site of action of drugs influencing acid secretion.
The initial step in controlling physiological secretion is the release of gastrin from G cells. This acts through its CCK2 receptor on ECL cells to release histamine and may also have a secondary direct effect on parietal cells themselves, although this is not entirely clear. Histamine acts on parietal cell H2 receptors to elevate cAMP that activates the secretion of acid by the proton pump. Direct vagal stimulation also provokes acid secretion and released acetylcholine directly stimulates M3 receptors on parietal cells. Somatostatin probably exerts a tonic inhibitory influence on G cells, ECL cells and parietal cells, while local (or therapeutically administered) prostaglandins exert inhibitory effects predominately on ECL cell function. AA, arachidonic acid; ACh, acetylcholine; C, symport carrier for K+ and Cl−; CCK2, gastrin/cholecystokinin receptor; ECL, mast cell-like histamine-secreting enterochromaffin cell; NSAIDs, non-steroidal anti-inflammatory drugs; P, proton pump (H+-K+-ATPase); PGE2, prostaglandin E2.
Gastrin also stimulates histamine synthesis by ECL cells and indirectly increases pepsinogen secretion, stimulates blood flow and increases gastric motility. Release of this hormone is controlled by both neuronal transmitters and blood-borne mediators, as well as by the chemistry of the stomach contents. Amino acids and small peptides directly stimulate the gastrin-secreting cells, as do milk and solutions of calcium salts, explaining why it is inappropriate to use calcium-containing salts as antacids.
Acetylcholine
Acetylcholine (together with a battery of other neurotransmitters and peptides), released from postganglionic cholinergic neurons, stimulates specific muscarinic M3 receptors on the surface of the parietal cells (see Ch. 13), thereby elevating intracellular Ca2+ and stimulating the release of protons. It also has complex effects on other cell types; by inhibiting somatostatin release from D cells, it potentiates its action on parietal cell acid secretion.
Prostaglandins
Most cells of the gastrointestinal tract produce prostaglandins (PGs; see Ch. 6), the most important being PGE2 and I2. Prostaglandins exert ‘cytoprotective’ effects on many aspects of gastric function including increasing bicarbonate secretion (EP1/2 receptors), increasing the release of protective mucin (EP4 receptor), reducing gastric acid output probably by acting on EP2/3 receptors on ECL cells and preventing the vasoconstriction (and thus damage to the mucosa) that follows injury or insult. This is probably an action mediated through EP2/4 receptors. Misoprostol (see below) is a synthetic prostaglandin that probably exploits many of these effects to bring about its therapeutic action.
The Coordination of Factors Regulating Acid Secretion
The regulation of the parietal cell is complex and many local hormones probably play a role in the fine-tuning of the secretory response. The generally accepted model today is that the gastrin–ECL–parietal cell axis is the dominant mechanism for controlling acid secretion. According to this idea (see Fig. 29.2), which is supported by the majority of transgenic ‘knockout’ mouse studies, the initial step in controlling physiological secretion is the release of gastrin from G cells. This acts through its CCK2 receptor on ECL cells to release histamine and may also have a secondary direct effect on parietal cells themselves, although this has been disputed. Histamine acts on H2 receptors on parietal cells to elevate cAMP and to activate the secretion of protons as described.
Direct vagal stimulation can also provoke acid secretion (the basis for ‘stress ulcers’) through a release of acetylcholine, which directly stimulates M3 receptors on parietal cells. Somatostatin probably exerts a tonic inhibitory influence on G cells, ECL and parietal cells, and local (or therapeutically administered) prostaglandins, acting through EP2/3 receptors, exert inhibitory effects predominantly on ECL cell function.
This control system is clearly complex but prolonged exposure of tissues to excess acid secretion is dangerous and must be tightly regulated (see Schubert & Peura, 2008).

Drugs Used to Inhibit or Neutralise Gastric Acid Secretion
The principal clinical indications for reducing acid secretion are peptic ulceration (both duodenal and gastric), GORD (in which gastric juice causes damage to the oesophagus) and the Zollinger–Ellison syndrome (a rare condition that is caused by a gastrin-producing tumour).
The reasons why peptic ulcers develop are not fully understood, although infection of the stomach mucosa with Helicobacter pylori2—a Gram-negative bacillus that causes chronic gastritis—is now generally considered to be a major cause (especially of duodenal ulcer) and, while there are some problems with this notion (see Axon, 2007), forms the usual basis for therapy. Treatment of H. pylori infection is discussed below.
Many non-specific NSAIDs (see Ch. 26) cause gastric bleeding and erosions by inhibiting cyclo-oxygenase-1, the enzyme responsible for synthesis of protective prostaglandins (see above). More selective cyclo-oxygenase-2 inhibitors such as celecoxib appear to cause less stomach damage (but see Ch. 26 for a discussion of this issue).
Therapy of peptic ulcer and reflux oesophagitis aims to decrease the secretion of gastric acid with H2 receptor antagonists or proton pump inhibitors, and/or to neutralise secreted acid with antacids (see Huang & Hunt, 2001). These treatments are often coupled with measures to eradicate H. pylori (see Horn, 2000).
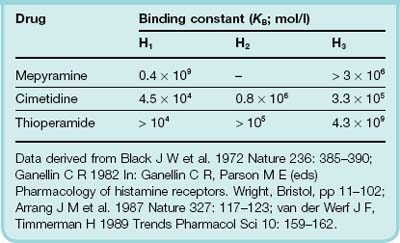
Histamine H2 Receptor Antagonists
The discovery and development of histamine H2-blocking drugs by Black and his colleagues was a major breakthrough in the treatment of gastric ulcers—a condition that could hitherto only be treated by (sometimes rather heroic) surgery.3 The ability to distinguish between histamine receptor subtypes using pharmacological agents was, in itself, a major intellectual advance (see Table 29.1). H2 receptor antagonists competitively inhibit histamine actions at all H2 receptors, but their main clinical use is as inhibitors of gastric acid secretion. They can inhibit histamine- and gastrin-stimulated acid secretion; pepsin secretion also falls with the reduction in volume of gastric juice. These agents not only decrease both basal and food-stimulated acid secretion by 90% or more, but numerous clinical trials indicate that they also promote healing of gastric and duodenal ulcers. However, relapses are likely to follow after cessation of treatment.
The drugs used are cimetidine, ranitidine (sometimes in combination with bismuth; see below), nizatidine and famotidine. There is little difference between them. The effect of cimetidine on gastric secretion in human subjects is shown in Figure 29.3. The clinical use of H2 receptor antagonists is given in the clinical box.
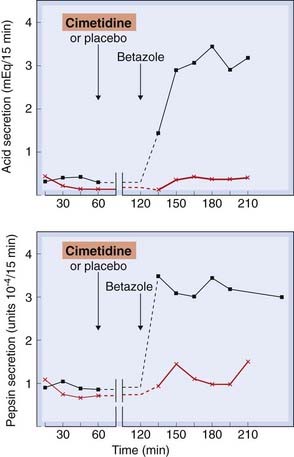
Fig. 29.3 The effect of cimetidine on betazole-stimulated gastric acid and pepsin secretion in humans.
Either cimetidine or a placebo was given orally 60 min prior to a subcutaneous injection (1.5 mg/kg) of betazole, a relatively specific histamine H2-receptor agonist that stimulates gastric acid secretion.
(Modified from Binder H J, Donaldson R M 1978 Gastroenterology 74: 371–375.)
Pharmacokinetic aspects and unwanted effects
The drugs are generally given orally and are well absorbed, although preparations for intramuscular and intravenous use are also available (except famotidine). Dosage regimens vary depending on the condition under treatment. Low-dosage over-the-counter formulations of cimetidine, ranitidine and famotidine are available for short-term uses, without prescription, from pharmacies.
Unwanted effects are rare. Diarrhoea, dizziness, muscle pains, alopecia, transient rashes, confusion in the elderly and hypergastrinaemia have been reported. Cimetidine sometimes causes gynaecomastia in men and, rarely, a decrease in sexual function. This is probably caused by a modest affinity for androgen receptors. Cimetidine (but not other H2 receptor antagonists) also inhibits cytochrome P450, and can retard the metabolism (and thus potentiate the action) of a range of drugs including oral anticoagulants and tricyclic antidepressants.
Proton Pump Inhibitors
The first proton pump inhibitor was omeprazole, which irreversibly inhibits the H+-K+-ATPase (the proton pump), the terminal step in the acid secretory pathway (see Figs 29.1 and 29.2). Both basal and stimulated gastric acid secretion (Fig. 29.4) are reduced. The drug is a weak base, and accumulates in the acid environment of the canaliculi of the stimulated parietal cell where it is activated. This preferential accumulation means that it has a specific effect on these cells. Other proton pump inhibitors (all of which are very similar) include esomeprazole (the [S] isomer of omeprazole), lansoprazole, pantoprazole and rabeprazole. The clinical use of these inhibitors is given in the clinical box.
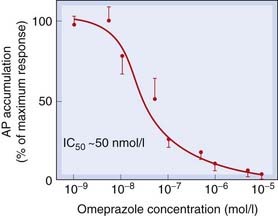
Fig. 29.4 The inhibitory action of omeprazole on acid secretion from isolated human gastric glands stimulated by 50 µmol/l histamine.
Acid secretion was measured by the accumulation of a radiolabelled weak base, aminopyrine (AP), in the secretory channels. The data represent the mean and standard error of measurements from eight patients.
(Adapted from Lindberg P et al. 1987 Trends Pharmacol Sci 8: 399–402.)
Pharmacokinetic aspects and unwanted effects
Oral administration is the most common route of administration, although some injectable preparations are available. Omeprazole is given orally, but as it degrades rapidly at low pH, it is administered as capsules containing enteric-coated granules. It is absorbed and, from the blood, passes into the parietal cells and then into the canaliculi. Increased doses give disproportionately higher increases in plasma concentration (possibly because its inhibitory effect on acid secretion improves its own bioavailability). Although its half-life is about 1 h, a single daily dose affects acid secretion for 2–3 days, because it accumulates in the canaliculi and inhibits H+-K+-ATPase irreversibly. With daily dosage, there is an increasing antisecretory effect for up to 5 days, after which a plateau is reached.
Unwanted effects of this class of drugs are uncommon. They may include headache, diarrhoea (both sometimes severe) and rashes. Dizziness, somnolence, mental confusion, impotence, gynaecomastia, and pain in muscles and joints have been reported. Proton pump inhibitors should be used with caution in patients with liver disease, or in women who are pregnant or breastfeeding. The use of these drugs may ‘mask’ the symptoms of gastric cancer.
Antacids
Antacids are the simplest way to treat the symptoms of excessive gastric acid secretion. They directly neutralise acid, which also has the effect of inhibiting the activity of peptic enzymes, which practically ceases at pH 5. Given in sufficient quantity for long enough, they can produce healing of duodenal ulcers but are less effective for gastric ulcers.
Most antacids in common use are salts of magnesium and aluminium. Magnesium salts cause diarrhoea and aluminium salts constipation, so mixtures of these two can, happily, be used to preserve normal bowel function. Some preparations of these substances (e.g. magnesium trisilicate mixtures and some proprietary aluminium preparations) contain high concentrations of sodium and should not be given to patients on a sodium-restricted diet. Numerous antacid preparations are available; a few of the more significant are given below.
Magnesium hydroxide is an insoluble powder that forms magnesium chloride in the stomach. It does not produce systemic alkalosis, because Mg2+ is poorly absorbed from the gut. Another salt, magnesium trisilicate, is an insoluble powder that reacts slowly with the gastric juice, forming magnesium chloride and colloidal silica. This agent has a prolonged antacid effect, and it also adsorbs pepsin.
Aluminium hydroxide gel forms aluminium chloride in the stomach; when this reaches the intestine, the chloride is released and is reabsorbed. Aluminium hydroxide raises the pH of the gastric juice to about 4, and also adsorbs pepsin. Its action is gradual, and its effect continues for several hours.4 Colloidal aluminium hydroxide combines with phosphates in the gastrointestinal tract, and the increased excretion of phosphate in the faeces that occurs results in decreased excretion of phosphate via the kidney. This effect has been used in treating patients with chronic renal failure (see Ch. 28).
Alginates or simeticone are sometimes combined with antacids. Alginates are believed to increase the viscosity and adherence of mucus to the oesophageal mucosa, forming a protective barrier (see also below), whereas simeticone is an anti-foaming agent, intended to relieve bloating and flatulence.
Treatment of Helicobactor Pylori Infection
H. pylori infection has been implicated as a causative factor in the production of gastric and, more particularly, duodenal ulcers, as well as a risk factor for gastric cancer. Indeed, some would argue that infectious gastroduodenitis is actually the chief clinical entity associated with ulcers, and gastric cancer its prominent sequela. Certainly, eradication of H. pylori infection promotes rapid and long-term healing of ulcers, and it is routine practice to test for the organism in patients presenting with suggestive symptoms. If the test is positive, then the organism can generally be eradicated with a 1- or 2-week regimen of ‘triple therapy’, comprising a proton pump inhibitor in combination with the antibacterials amoxicillin and metronidazole or clarithromycin (see Ch. 50); other combinations are also used. Bismuth-containing preparations (see below) are sometimes added. While elimination of the bacillus can produce long-term remission of ulcers, reinfection with the organism can occur.
Drugs That Protect the Mucosa
Some agents, termed cytoprotective, are said to enhance endogenous mucosal protection mechanisms (see above) and/or to provide a physical barrier over the surface of the ulcer.
Bismuth chelate
Bismuth chelate (colloidal bismuth subcitrate, tripotassium dicitratobismuthate) is used in combination regimens to treat H. pylori. It has toxic effects on the bacillus, and may also prevent its adherence to the mucosa or inhibit its bacterial proteolytic enzymes. It is also believed to have other mucosa-protecting actions, by mechanisms that are unclear, and is widely used as an over-the-counter remedy for mild gastrointestinal symptoms.Very little is absorbed, but if renal excretion is impaired, the raised plasma concentrations of bismuth can result in encephalopathy.
Unwanted effects include nausea and vomiting, and blackening of the tongue and faeces.
Sucralfate
Sucralfate is a complex of aluminium hydroxide and sulfated sucrose, which releases aluminium in the presence of acid. The residual complex carries a strong negative charge and binds to cationic groups in proteins, glycoproteins, etc. It can form complex gels with mucus, an action that is thought to decrease the degradation of mucus by pepsin and to limit the diffusion of H+. Sucralfate can also inhibit the action of pepsin and stimulate secretion of mucus, bicarbonate and prostaglandins from the gastric mucosa. All these actions contribute to its mucosa-protecting action.
Sucralfate is given orally, and in the acid environment of the stomach the polymerised product forms a tenacious paste, which can produce an obstructive lump (known as a bezoar5) that gets stuck in the stomach; about 30% is still present in the stomach 3 h after administration. It reduces the absorption of a number of other drugs, including fluoroquinolone antibiotics, theophylline, tetracycline, digoxin and amitriptyline. Because it requires an acid environment for activation, antacids given concurrently or prior to its administration will reduce its efficacy.
Unwanted effects are few, the most common being constipation. Less common effects include dry mouth, nausea, vomiting, headache, bezoar formation and rashes.
Misoprostol
Prostaglandins of the E and I series have a generally protective action in the gastrointestinal tract, and a deficiency in endogenous production (after ingestion of a NSAID, for example) may contribute to ulcer formation. Misoprostol is a stable analogue of prostaglandin E1. It is given orally and is used to promote the healing of ulcers or to prevent the gastric damage that can occur with chronic use of NSAIDs. It exerts a direct action on the ECL cell (and possibly parietal cell also; Fig. 29.2), inhibiting the basal secretion of gastric acid as well as the stimulation of production seen in response to food, pentagastrin and caffeine. It also increases mucosal blood flow and augments the secretion of mucus and bicarbonate.
Unwanted effects include diarrhoea and abdominal cramps; uterine contractions can also occur, so the drug should not be given during pregnancy (unless deliberately to induce a therapeutic abortion; see Ch. 34). Prostaglandins and NSAIDs are discussed more fully in Chs 6 and 26.
Vomiting
Nausea and vomiting are unwanted side effects of many clinically used drugs, notably those used for cancer chemotherapy as well as opioids, general anaesthetics and digoxin. They also occur in motion sickness,6 during early pregnancy and in numerous disease states (e.g. migraine) as well as bacterial and viral infections.
The Reflex Mechanism of Vomiting
Vomiting is regulated centrally by the vomiting centre and the chemoreceptor trigger zone (CTZ), both of which lie in the medulla. The CTZ is sensitive to chemical stimuli and is the main site of action of many emetic and antiemetic drugs. The blood–brain barrier in the neighbourhood of the CTZ is relatively permeable, allowing circulating mediators to act directly on this centre. The CTZ also regulates motion sickness. Impulses from the CTZ pass to those areas of the brain stem—known collectively as the vomiting centre—that control and integrate the visceral and somatic functions involved in vomiting.
An outline of the pathways involved in the control of vomiting is given in Figure 29.5 and reviewed in detail by Hornby (2001). The main neurotransmitters are acetylcholine, histamine, 5-hydroxytryptamine (5-HT), dopamine and substance P, and receptors for these transmitters have been demonstrated in the relevant areas (see Chs 12–14 and 38). It has been hypothesised that enkephalins (see Chs 19 and 41) are also implicated in the mediation of vomiting, acting possibly at δ (CTZ) or µ (vomiting centre) opioid receptors. Substance P (see Ch. 19) acting at neurokinin-1 receptors in the CTZ, and endocannabinoids (Ch. 18), may also be involved.
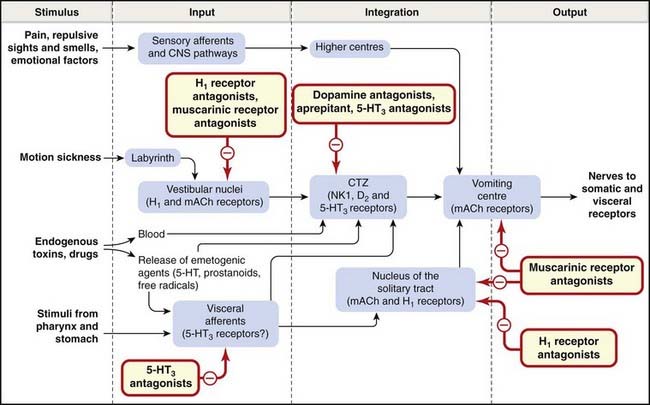
Fig. 29.5 Schematic diagram of the factors involved in the control of vomiting, with the probable sites of action of antiemetic drugs.
The cerebellum may function as a second relay or gating mechanism in the link between the labyrinth and chemoreceptor trigger zone (CTZ; not shown). 5-HT3, 5-hydroxytryptamine type 3; D2, dopamine D2; H1, histamine H1; mACh, muscarinic acetylcholine; NK1, neurokinin 1.
(Based partly on a diagram from Borison H L et al. 1981 J Clin Pharmacol 21: 235–295.)
The neurobiology of nausea is much less well understood. Nausea and vomiting may occur together or separately and may subserve different physiological functions (see Andrews & Horn, 2006). From the pharmacologist’s viewpoint, it is easier to control vomiting than nausea, and many effective antiemetics (e.g. 5-HT3 antagonists) are much less successful in this regard.
Antiemetic Drugs
Several antiemetic agents are available, and these are generally used for specific conditions, although there may be some overlap. Such drugs are of particular importance as an adjunct to cancer chemotherapy, where the nausea and vomiting produced by many cytotoxic drug (see Ch. 55) can be almost unendurable.7 In using drugs to treat the morning sickness of pregnancy, the problem of potential damage to the fetus has always to be borne in mind. In general, all drugs should be avoided during the first 3 months of pregnancy, if possible. Details of the main categories of antiemetics are given below, and their main clinical uses are summarised in the box.
Receptor Antagonists
Many H1 (see Ch. 26), muscarinic (see Ch. 13), 5-HT3 (see Ch. 15) and dopamine (Ch. 45) receptor antagonists exhibit clinically useful antiemetic activity.
H1 receptor antagonists
Cinnarizine, cyclizine and promethazine are the most commonly employed; they are effective against nausea and vomiting arising from many causes, including motion sickness and the presence of irritants in the stomach. None is very effective against substances that act directly on the CTZ. Promethazine is used for morning sickness of pregnancy (on the rare occasions when this is so severe that drug treatment is justified), and has been used by NASA to treat space motion sickness. Drowsiness and sedation, while possibly contributing to their clinical efficacy, are the chief unwanted effects.
Muscarinic receptor antagonists
Hyoscine (scopolamine) is employed principally for prophylaxis and treatment of motion sickness, and may be administered orally or as a transdermal patch. Dry mouth and blurred vision are the most common unwanted effects. Drowsiness also occurs, but the drug has less sedative action than the antihistamines because of poor central nervous system penetration.
5-HT3 receptor antagonists
Dolasetron, granisetron, ondansetron, palonosteron and tropisetron (see Ch. 15), are of particular value in preventing and treating the vomiting and, to a lesser extent the nausea, commonly encountered postoperatively or that caused by radiation therapy or administration of cytotoxic drugs such as cisplatin. The primary site of action of these drugs is the CTZ. They may be given orally or by injection (sometimes helpful if nausea is already present). Unwanted effects such as headache and gastrointestinal upsets are relatively uncommon.
Dopamine antagonists
Antipsychotic phenothiazines (see Ch. 45), such as chlorpromazine, perphenazine, prochlorperazine and trifluoperazine, are effective antiemetics commonly used for treating the more severe nausea and vomiting associated with cancer, radiation therapy, cytotoxic drugs, opioids, anaesthetics and other drugs. They can be administered orally, intravenously or by suppository. They act mainly as antagonists of the dopamine D2 receptors in the CTZ (see Fig. 29.5) but they also block histamine and muscarinic receptors.
Unwanted effects are common and include sedation (especially chlorpromazine), hypotension and extrapyramidal symptoms including dystonias and tardive dyskinesia (Ch. 45).
Other antipsychotics, such as haloperidol and levomepromazine (Ch. 45), also act as D2 antagonists in the CTZ and can be used for acute chemotherapy-induced emesis.
Metoclopramide and domperidone
Metoclopramide is a D2 receptor antagonist (Fig. 29.5), closely related to the phenothiazine group, that acts centrally on the CTZ and also has a peripheral action on the gastrointestinal tract itself, increasing the motility of the oesophagus, stomach and intestine. This not only adds to the antiemetic effect, but explains its use in the treatment of gastro-oesophageal reflux (see below) and hepatic and biliary disorders. As metoclopramide also blocks dopamine receptors (see Ch. 43) elsewhere in the central nervous system, it produces a number of unwanted effects including disorders of movement (more common in children and young adults), fatigue, motor restlessness, spasmodic torticollis (involuntary twisting of the neck) and occulogyric crises (involuntary upward eye movements). It stimulates prolactin release (see Ch. 32), causing galactorrhoea and disorders of menstruation.
Domperidone is a similar drug often used to treat vomiting due to cytotoxic therapy as well as gastrointestinal symptoms. Unlike metoclopramide, it does not readily penetrate the blood–brain barrier and is consequently less prone to producing central side effects. Both drugs are given orally, have plasma half-lives of 4–5 h and are excreted in the urine.
NK1 receptor antagonists
Aprepitant blocks substance P (NK1) receptors (see Ch. 19) in the CTZ and vomiting centre. Substance P causes vomiting when injected intravenously and is released by gastrointestinal vagal afferent nerves as well as in the vomiting centre itself. Aprepitant is given orally, and is effective in controlling the late phase of emesis caused by cytotoxic drugs, with few significant unwanted effects. Fosaprepitant is a prodrug of aprepitant, given intravenously.
Other Antiemetic Drugs
Anecdotal evidence originally suggested the possibility of using cannabinoids (see Ch. 18) as antiemetics (see Pertwee, 2001). The synthetic cannabinol nabilone has been found to decrease vomiting caused by agents that stimulate the CTZ, and is sometimes effective where other drugs have failed (see Ch. 18). The antiemetic effect is antagonised by naloxone, which implies that opioid receptors may be important in the mechanism of action. Nabilone is given orally; it is well absorbed from the gastrointestinal tract and is metabolised in many tissues. Its plasma half-life is approximately 120 min, and its metabolites are excreted in the urine and faeces.
Unwanted effects are common, especially drowsiness, dizziness and dry mouth. Mood changes and postural hypotension are also fairly frequent. Some patients experience hallucinations and psychotic reactions, resembling the effect of other cannabinoids (see Ch. 18).
High-dose glucocorticoids (particularly dexamethasone; see Chs 26 and 32) can also control emesis, especially when this is caused by cytotoxic drugs. The mechanism of action is not clear. Dexamethasone can be used alone but is frequently deployed in combination with a phenothiazine, ondansetron or aprepitant.
Clinical use of antiemetic drugs 
The Motility of the Gastrointestinal Tract
Drugs that alter the motility of the gastrointestinal tract include:
Purgatives
The transit of food through the intestine may be hastened by several different types of drugs, including laxatives, faecal softeners and stimulant purgatives. The latter agents may be used to relieve constipation or to clear the bowel prior to surgery or examination.
Bulk and Osmotic Laxatives
The bulk laxatives include methylcellulose and certain plant extracts such as sterculia, agar, bran and ispaghula husk. These agents are polysaccharide polymers that are not digested in the upper part of the gastrointestinal tract. They form a bulky hydrated mass in the gut lumen promoting peristalsis and improving faecal consistency. They may take several days to work but have no serious unwanted effects.
The osmotic laxatives consist of poorly absorbed solutes—the saline purgatives—and lactulose. The main salts in use are magnesium sulfate and magnesium hydroxide. By producing an osmotic load, these agents trap increased volumes of fluid in the lumen of the bowel, accelerating the transfer of the gut contents through the small intestine. This results in an abnormally large volume entering the colon, causing distension and purgation within about an hour. Abdominal cramps can occur. The amount of magnesium absorbed after an oral dose is usually too small to have adverse systemic effects, but these salts should be avoided in small children and in patients with poor renal function, in whom they can cause heart block, neuromuscular block or central nervous system depression. While isotonic or hypotonic solutions of saline purgatives cause purgation, hypertonic solutions can cause vomiting. Sometimes, other sodium salts of phosphate and citrate are given rectally, by suppository, to relieve constipation.
Lactulose is a semisynthetic disaccharide of fructose and galactose. It is poorly absorbed and produces an effect similar to that of the other osmotic laxatives. It takes 2–3 days to act. Unwanted effects, seen with high doses, include flatulence, cramps, diarrhoea and electrolyte disturbance. Tolerance can develop. Another agent, macrogols, which consists of inert ethylene glycol polymers, acts in the same way.
Faecal Softeners
Docusate sodium is a surface-active compound that acts in the gastrointestinal tract in a manner similar to a detergent and produces softer faeces. It is also a weak stimulant laxative. Other agents that achieve the same effect include arachis oil, which is given as an enema, and liquid paraffin, although this is now seldom used.
Stimulant Laxatives
The stimulant laxative drugs act mainly by increasing electrolyte and hence water secretion by the mucosa, and also by increasing peristalsis—possibly by stimulating enteric nerves. Abdominal cramping may be experienced as a side effect with almost any of these drugs.
Bisacodyl may be given by mouth but is often given by suppository. In the latter case, it stimulates the rectal mucosa, inducing defaecation in 15–30 min. Glycerol suppositories act in the same manner. Sodium picosulfate and docusate sodium have similar actions. The former is given orally and is often used in preparation for intestinal surgery or colonoscopy.
Senna and dantron are anthroquinone laxatives. The active principle (after hydrolysis of glycosidic linkages in the case of the plant extract, senna) directly stimulates the myenteric plexus, resulting in increased peristalsis and thus defaecation. Dantron is similar. As this drug is a skin irritant and may be carcinogenic, it is generally used only in the terminally ill.
Laxatives of any type should not be used when there is obstruction of the bowel. Overuse can lead to an atonic colon where the natural propulsive activity is diminished. In these circumstances, the only way to achieve defaecation is to take further amounts of laxatives, so a sort of dependency arises.
Drugs That Increase Gastrointestinal Motility
Domperidone is primarily used as an antiemetic (as described above), but it also increases gastrointestinal motility (although the mechanism is unknown). Clinically, it increases lower oesophageal sphincter pressure (thus inhibiting gastro-oesophageal reflux), increases gastric emptying and enhances duodenal peristalsis. It is useful in disorders of gastric emptying and in chronic gastric reflux.
Metoclopramide (also an antiemetic; see above) stimulates gastric motility, causing a marked acceleration of gastric emptying. It is useful in gastro-oesophageal reflux and in disorders of gastric emptying, but is ineffective in paralytic ileus.
Now withdrawn (because it precipitated fatal cardiac arrhythmias), cisapride stimulates acetylcholine release in the myenteric plexus in the upper gastrointestinal tract through a 5-HT4 receptor-mediated effect. Tegaserod (also recently withdrawn on account of suspected increase in heart attacks and strokes) acts similarly. These drugs raise oesophageal sphincter pressure and increase gut motility. They were used for treating reflux oesophagitis and in disorders of gastric emptying.
Antidiarrhoeal Agents
There are numerous causes of diarrhoea, including underlying disease, infection, toxins and even anxiety. It may also arise as a side effect of drug or radiation therapy. Repercussions range from mild discomfort and inconvenience to a medical emergency requiring hospitalisation and parenteral fluid and electrolyte replacement therapy. Globally, acute diarrhoeal disease is one of the principal causes of death in malnourished infants, especially in developing countries where medical care is less accessible and 1–2 million children die each year for want of simple counter-measures.
During an episode of diarrhoea, there is an increase in the motility of the gastrointestinal tract, accompanied by an increased secretion coupled with a decreased absorption of fluid, which leads to a loss of electrolytes (particularly Na+) and water. Cholera toxins and some other bacterial toxins produce a profound increase in electrolyte and fluid secretion by irreversibly activating the G-proteins that couple the surface receptors of the mucosal cells to adenylyl cyclase (see Ch. 3).
There are three approaches to the treatment of severe acute diarrhoea:
The maintenance of fluid and electrolyte balance by means of oral rehydration is the first priority, and wider application of this cheap and simple remedy could save the lives of many infants in the developing world. Many patients require no other treatment. In the ileum, as in parts of the nephron, there is co-transport of Na+ and glucose across the epithelial cell. The presence of glucose (and some amino acids) therefore enhances Na+ absorption and thus water uptake. Preparations of sodium chloride and glucose for oral rehydration are available in powder form, ready to be dissolved in water before use.
Many gastrointestinal infections are viral in origin, and because those that are bacterial generally resolve fairly rapidly, the use of anti-infective agents is usually neither necessary nor useful. Other cases may require more aggressive therapy, however. Campylobacter sp. is the commonest cause of bacterial gastroenteritis in the UK, and severe infections may require ciprofloxacin (Ch. 50). The most common bacterial organisms encountered by travellers include Escherichia coli, Salmonella and Shigella, as well as protozoa such as Giardia and Cryptosporidium spp. Drug treatment (Chs 50 and 53) may be necessary in these and other more serious infections.
Traveller’s Diarrhoea
More than 3 million people cross international borders each year. Many travel hopefully, but some 20–50% come back ill, having encountered enterotoxin-producing E. coli (the most common cause) or other organisms. Most infections are mild and self-limiting, requiring only oral replacement of fluid and salt, as detailed above. General principles for the drug treatment of traveller’s diarrhoea are detailed by Gorbach (1987).8 Up-to-date information on the condition, including the prevalence of infectious organisms around the globe as well as recommended treatment guidelines, is issued in the UK by the National Travel Health Network and Centre (see Web links in the reference list).
Antimotility and Spasmolytic Agents
The main pharmacological agents that decrease motility are opiates (Ch. 41) and muscarinic receptor antagonists (Ch. 13). Agents in this latter group are seldom employed as primary therapy for diarrhoea because of their actions on other systems, but small doses of atropine are sometimes used, combined with diphenoxylate (see below). The action of morphine, the archetypal opiate, on the alimentary tract is complex; it increases the tone and rhythmic contractions of the intestine but diminishes propulsive activity. The pyloric, ileocolic and anal sphincters are contracted, and the tone of the large intestine is markedly increased. Its overall effect is constipating.
The main opiates used for the symptomatic relief of diarrhoea are codeine (a morphine congener), diphenoxylate and loperamide (both pethidine congeners that do not readily penetrate the blood–brain barrier and are used only for their actions in the gut). All may have unwanted effects including constipation, abdominal cramps, drowsiness and dizziness. Paralytic ileus can also occur. They should not be used in young (< 4 years of age) children.
Loperamide is the drug of first choice for traveller’s diarrhoea and is a component of several proprietary antidiarrhoeal medicines. It has a relatively selective action on the gastrointestinal tract and undergoes significant enterohepatic cycling. It reduces the frequency of abdominal cramps, decreases the passage of faeces and shortens the duration of the illness.
Diphenoxylate also lacks morphine-like activity in the central nervous system, although large doses (25-fold higher) produce typical opioid effects. Preparations of diphenoxylate usually contain atropine as well. Codeine and loperamide have antisecretory actions in addition to their effects on intestinal motility. Cannabinoid receptor agonists also reduce gut motility in animals, most probably by decreasing acetylcholine release from enteric nerves. There have been anecdotal reports of a beneficial effect of cannabis against dysentery and cholera.
Drugs that reduce spasm in the gut are also of value in irritable bowel syndrome and diverticular disease. Muscarinic receptor antagonists (Ch. 13) used for this purpose include atropine, hyoscine, propantheline and dicycloverine. The last named is thought to have some additional direct relaxant action on smooth muscle. All produce antimuscarinic side effects such as dry mouth, blurred vision and urinary retention. Mebeverine, a derivative of reserpine, has a direct relaxant action on gastrointestinal smooth muscle. Unwanted effects are few.
Adsorbents
Adsorbent agents are used extensively in the symptomatic treatment of diarrhoea, although properly controlled trials proving efficacy have not been carried out. The main preparations used contain kaolin, pectin, chalk, charcoal, methylcellulose and activated attapulgite (magnesium aluminium silicate). It has been suggested that these agents may act by adsorbing microorganisms or toxins, by altering the intestinal flora or by coating and protecting the intestinal mucosa, but there is no hard evidence for this. They are often given as mixtures with other drugs (e.g. kaolin and morphine mixture BP).
Drugs for Chronic Bowel Disease
This category comprises irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD). IBS is characterised by bouts of diarrhoea, constipation or abdominal pain. The aetiology of the disease is uncertain, but psychological factors may play a part. Treatment is symptomatic, with a high-residue diet plus loperamide or a laxative if needed.
Ulcerative colitis and Crohn’s disease are forms of IBD, affecting the colon or ileum. They are autoimmune inflammatory disorders, which can be severe and progressive, requiring long-term drug treatment with anti-inflammatory and immunosuppressant drugs (see Ch. 26), and occasionally surgical resection. The following agents are used.
Glucocorticoids
Glucocorticoids are potent anti-inflammatory agents and are dealt with fully in Chapters 26 and 32. The drugs of choice are generally prednisolone or budesonide (although others can be used), given orally or locally into the bowel by suppository or enema.
Aminosalicylates
While glucocorticoids are useful for the acute attacks of inflammatory bowel diseases, they are not the ideal for the long-term treatment (because of their side effects). Maintenance of remission in both ulcerative colitis and Crohn’s disease is generally achieved with aminosalicylates, although they are less useful in the latter condition.
Sulfasalazine consists of the sulfonamide sulfapyridine linked to 5-aminosalicylic acid (5-ASA). The latter forms the active moiety when it is released in the colon. Its mechanism of action is obscure. It may reduce inflammation by scavenging free radicals, by inhibiting prostaglandin and leukotriene production, and/or by decreasing neutrophil chemotaxis and superoxide generation. Its unwanted effects are diarrhoea, salicylate sensitivity and interstitial nephritis. 5-ASA is not absorbed, but the sulfapyridine moiety, which seems to be therapeutically inert in this instance, is absorbed, and its unwanted effects are those associated with the sulfonamides (see Ch. 50).
Newer compounds in this class, which presumably share a similar mechanism of action, include mesalazine (5-ASA itself), olsalazine (a 5-ASA dimer linked by a bond that is hydrolysed by colonic bacteria) and balsalazide (a prodrug from which 5-ASA is also released following hydrolysis of a diazo linkage).
Other Drugs
The immunosuppressants azathioprine and 6-mercapto-purine (see Ch. 26) are also sometimes used in patients with severe disease. Recently, infliximab and adalimumab, monoclonal antibodies directed against tumour necrosis factor (TNF)-α, (see Ch. 26) have been used with success for the treatment of inflammatory bowel diseases. These drugs are expensive, and in the UK their use is restricted to moderate/severe Crohn’s disease that is unresponsive to glucocorticoids or immunomodulators. The antiallergy drug sodium cromoglicate (see Ch. 27) is sometimes used for treating gastrointestinal symptoms associated with food allergies.
Drugs Affecting the Biliary System
The commonest pathological condition of the biliary tract is cholesterol cholelithiasis, i.e. the formation of gallstones with high cholesterol content. Surgery is generally the preferred option, but there are orally active drugs that dissolve non-calcified ‘radiolucent’ cholesterol gallstones. The principal agent is ursodeoxycholic acid, a minor constituent of human bile (but the main bile acid in the bear, hence urso-). Diarrhoea is the main unwanted effect.
Biliary colic, the pain produced by the passage of gallstones through the bile duct, can be very intense, and immediate relief may be required. Morphine relieves the pain effectively, but it may have an undesirable local effect because it constricts the sphincter of Oddi and raises the pressure in the bile duct. Buprenorphine may be preferable. Pethidine has similar actions, although it relaxes other smooth muscle, for example that of the ureter. Atropine is commonly employed to relieve biliary spasm because it has antispasmodic action and may be used in conjunction with morphine. Glyceryl trinitrate (see Ch. 21) can produce a marked fall of intrabiliary pressure and may be used to relieve biliary spasm.
Future Directions
It might be thought that the widespread availability of several different types of safe antisecretory drug would have satisfied the medical need for antiulcer therapies, but this is not so. Although the incidence of ulcers has dropped, thanks to these drugs, other diseases associated with excess acid production (GORD, NSAID-induced damage) are on the increase, at least in the ‘developed’ countries. The prospects for new types of histamine antagonist (e.g. H3 antagonists) are being explored as are antagonists at gastrin receptors. The most interesting new candidates though are the potassium competitive acid blocker, several of which are in various stages of clinical development. Potassium ions are exchanged for protons by the proton pump (see Fig. 29.1) and so potassium antagonists would represent an alternative modality for inhibiting the secretion of acid. This novel field, as well as other projects, are discussed by Mossner & Caca (2005).
References and Further Reading
Black J.W., Duncan W.A.M., Durant C.J., et al. Definition and antagonism of histamine H2-receptors. Nature. 1972;236:385-390. (Seminal paper outlining the pharmacological approach to inhibition of acid secretion through antagonism at an alternative histamine receptor)
Innervation and hormones of the gastrointestinal tract
Hansen M.B. The enteric nervous system II: gastrointestinal functions. Pharmacol. Toxicol.. 2003;92:249-257. (Small review on the role of the enteric nervous system in the control of gastrointestinal motility, secretory activity, blood flow and immune status; easy to read)
Sanger G.J. Neurokinin NK1 and NK3 receptors as targets for drugs to treat gastrointestinal motility disorders and pain. Br. J. Pharmacol.. 2004;141:1303-1312. (Useful review that deals with the present and potential future uses of neurokinin antagonists in gastrointestinal physiology and pathology)
Spiller R. Serotonergic modulating drugs for functional gastrointestinal diseases. Br. J. Clin. Pharmacol.. 2002;54:11-20. (An excellent and ‘easily digestible’ article describing the latest thinking on the use of 5-hydroxytryptamine agonists and antagonists in gastrointestinal function; useful diagrams)
Chen D., Friis-Hansen L., Håkanson R., Zhao C.-M. Genetic dissection of the signaling pathways that control gastric acid secretion. Inflammopharmacology. 2005;13:201-207. (Describes experiments using receptor ‘knock outs’ to analyse the mechanisms that control gastric acid production)
Cui G., Waldum H.L. Physiological and clinical significance of enterochromaffin-like cell activation in the regulation of gastric acid secretion. World J. Gastroenterol.. 2007;13:493-496. (Short review on the central role of ECL cells in the regulation of gastric acid secretion. Easy to read)
Schubert M.L., Peura D.A. Control of gastric acid secretion in health and disease. Gastroenterology. 2008;134:1842-1860. (Excellent review of the physiology and pharmacology of gastric acid secretion. Authoritative and well illustrated)
Axon A.T. Relationship between Helicobacter pylori gastritis, gastric cancer and gastric acid secretion Adv. Med. Sci. 52:2007:55-60 (Takes a critical look at the epidemiological evidence for the relationship between H. pylori infection and gastric cancer)
Bateman D.N. Proton-pump inhibitors: three of a kind? Lancet. 1997;349:1637-1638. (Editorial commentary)
Blaser M.J. The bacteria behind ulcers. Sci. Am.. 1996;274:104-107. (Simple coverage, very good diagrams)
Blaser M.J. Helicobacter pylori and gastric disease. Br. Med. J.. 1998;316:1507-1510. (Succinct review; emphasis on future developments)
Horn J.H. The proton-pump inhibitors: similarities and differences. Clin. Ther.. 2000;22:266-280. (Excellent overview)
Huang J.Q., Hunt R.H. Pharmacological and pharmacodynamic essentials of H2-receptor antagonists and proton pump inhibitors for the practising physician. Baillière’s Best Pract. Res. Clin. Gastroenterol.. 2001;15:355-370.
Klotz U. The role of aminosalicylates at the beginning of the new millennium in the treatment of chronic inflammatory bowel disease. Eur. J. Clin. Pharmacol.. 2000;56:353-362.
Mossner J., Caca K. Developments in the inhibition of gastric acid secretion. Eur. J. Clin. Invest.. 2005;35:469-475. (Useful overview of several new directions in gastrointestinal drug development)
Pertwee R.G. Cannabinoids and the gastrointestinal tract. Gut. 2001;48:859-867.
Yeomans N.D., Tulassy Z., Juhász L., et al. A comparison of omeprazole with ranitidine for ulcers associated with nonsteroidal antiinflammatory drugs. N. Engl. J. Med.. 1998;338:719-726.
Andrews P.L., Horn C.C. Signals for nausea and emesis: implications for models of upper gastrointestinal diseases. Auton. Neurosci.. 2006;125:100-115.
Hesketh P.J. Potential role of the NK1 receptor antagonists in chemotherapy-induced nausea and vomiting. Support. Care Cancer. 2001;9:350-354.
Hornby P.J. Central neurocircuitry associated with emesis. Am. J. Med.. 2001;111:106S-112S. (Comprehensive review of central control of vomiting)
Tramèr M.R., Moore R., Reynolds D.J., McQuay H.J. A quantitative systematic review of ondansetron in treatment of established postoperative nausea and vomiting. Br. Med. J.. 1997;314:1088-1092.
Yates B.J., Miller A.D., Lucot J.B. Physiological basis and pharmacology of motion sickness: an update. Brain Res. Bull.. 1998;5:395-406. (Good account of the mechanisms underlying motion sickness and its treatment)
Motility of the gastrointestinal tract
De Las Casas C., Adachi J., Dupont H. Travellers’ diarrhoea. Aliment. Pharmacol. Ther.. 1999;13:1373-1378. (Review article)
Gorbach S.L. Bacterial diarrhoea and its treatment. Lancet. 1987;II:1378-1382.
http://www.nathnac.org http://www.nathnac.org (This is the site for the UK Health Protection Agency’s National Travel Health Network and Centre. There are two components to the site, one for lay people and one for health professionals. Click on the latter and enter ‘Travellers’ diarrhoea’ as a search term to retrieve current information and advice)
1Or GERD in the USA, to reflect the different spelling of esophageal.
2Helicobactor pylori infection in the stomach has been classified as a class 1 (definite) carcinogen for gastric cancer.
3This era has been referred to as the ‘BC’—before cimetidine—era of gastroenterology (Schubert & Peura 2008)! It is an indication of the clinical importance of the development of this drug.
4There was a suggestion—no longer widely believed—that aluminium could trigger Alzheimer’s disease. In fact, aluminium is not absorbed to any significant extent following oral administration of aluminium hydroxide, although when introduced by other routes (e.g. during renal dialysis with aluminium-contaminated solutions) it is extremely toxic.
5From the Persian word meaning ‘a cure for poisoning’. It refers to the belief that a concoction made from lumps of impacted rubbish retrieved from the stomach of goats would protect against poisoning by one’s enemies.
6In fact, the word nausea is derived from the Greek word meaning ‘boat’, with the obvious implication of associated motion sickness. Vomiting is derived from the Latin and a vomitorium was the ‘fast exit’ passageway in ancient theatres. It has a certain resonance, as we think you will agree!
7It was reported that a young, medically qualified patient being treated by combination chemotherapy for sarcoma stated that ‘the severity of vomiting at times made the thought of death seem like a welcome relief’.
8Who flippantly (although accurately) observed that ‘travel broadens the mind and loosens the bowels’.