20 Nitric oxide
Overview
Nitric oxide (NO) is a ubiquitous mediator with diverse functions. It is generated from L-arginine by nitric oxide synthase (NOS), an enzyme that occurs in endothelial, neuronal and inducible isoforms. In this chapter, we concentrate on general aspects of NO, especially its biosynthesis, degradation and effects. We touch on evidence that it can act as a circulating as well as a local mediator, and conclude with a brief consideration of the therapeutic potential of drugs that act on the L-arginine/NO pathway.
Introduction
Nitric oxide, a free radical gas, is formed in the atmosphere during lightning storms. Less dramatically, but with far-reaching biological consequences, it is also formed in an enzyme-catalysed reaction between molecular oxygen and L-arginine. The convergence of several lines of research led to the realisation that NO is a key signalling molecule in the cardiovascular and nervous systems, and that it has a role in host defence.
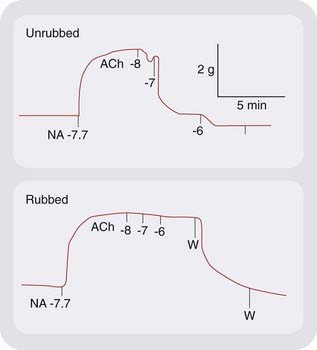
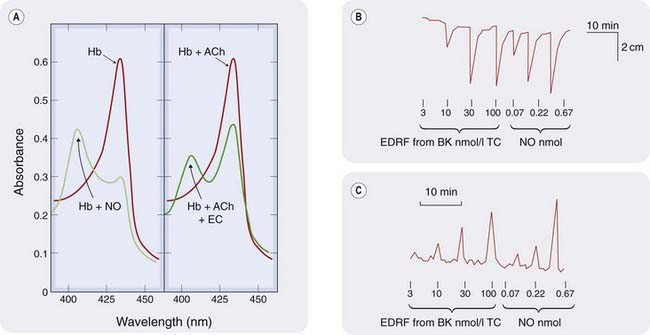
A physiological function of NO was discovered in the vasculature when it was shown that the endothelium-derived relaxing factor described by Furchgott & Zawadzki (1980) is NO (Figs 20.1 and 20.2). NO is the endogenous activator of soluble guanylyl cyclase, leading to the formation of cyclic GMP (cGMP), an important ‘second messenger’ (Ch. 3) in many cells, including nerves, smooth muscle, monocytes and platelets. Nitrogen and oxygen are neighbours in the periodic table, and NO shares several properties with O2, in particular a high affinity for haem and other iron–sulfur groups. This is important for activation of guanylyl cyclase, which contains a haem group, and for the inactivation of NO by haemoglobin (see below).
The role of NO in specific settings is described in other chapters: the endothelium in Chapter 22, the autonomic nervous system in Chapter 12, as a chemical transmitter and mediator of excitotoxicity in the central nervous system (CNS) in Chapters 36–38, and in the innate mediator-derived reactions of acute inflammation and the immune response in Chapter 17. Therapeutic uses of organic nitrates and of nitroprusside (NO donors) are described in Chapters 21 and 22.
Biosynthesis of Nitric Oxide and Its Control
Nitric oxide synthase (NOS) enzymes are central to the control of NO biosynthesis. There are three known isoforms: an inducible form (iNOS or NOS-II; expressed in macrophages and Kupffer cells, neutrophils, fibroblasts, vascular smooth muscle and endothelial cells in response to pathological stimuli such as invading microorganisms) and two so-called constitutive forms, which are present under physiological conditions in endothelium (eNOS or NOS-III) and in neurons (nNOS or NOS-I). eNOS is not restricted to endothelium. It is also present in cardiac myocytes, renal mesangial cells, osteoblasts and osteoclasts, airway epithelium and, in small amounts, platelets. The constitutive enzymes generate small amounts of NO, whereas iNOS produces much greater amounts both because of its high activity and because of its abundance, at least in pathological states associated with cytokine release.1
 All three NOS isoenzymes are dimers. They are structurally and functionally complex, bearing similarities to the cytochrome P450 enzymes (described in Ch. 9) that are so important in drug metabolism. Each isoform contains iron protoporphyrin IX (haem), flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN) and tetrahydrobiopterin (H4B) as bound prosthetic groups. They also bind L-arginine, reduced nicotinamide adenine dinucleotide phosphate (NADPH) and calcium–calmodulin. These prosthetic groups and ligands control the assembly of the enzyme into the active dimer. Calcium–calmodulin regulates electron transfer within the molecule.
All three NOS isoenzymes are dimers. They are structurally and functionally complex, bearing similarities to the cytochrome P450 enzymes (described in Ch. 9) that are so important in drug metabolism. Each isoform contains iron protoporphyrin IX (haem), flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN) and tetrahydrobiopterin (H4B) as bound prosthetic groups. They also bind L-arginine, reduced nicotinamide adenine dinucleotide phosphate (NADPH) and calcium–calmodulin. These prosthetic groups and ligands control the assembly of the enzyme into the active dimer. Calcium–calmodulin regulates electron transfer within the molecule.
Both nNOS and iNOS are soluble cytosolic enzymes, and eNOS is dually acylated by N-myristoylation and cysteine palmitoylation; these post-translational modifications lead to its association with membranes in the Golgi apparatus and in caveolae, specialised cholesterol-rich microdomains in the plasma membrane derived from the Golgi apparatus. In the caveolae, eNOS is associated with caveolin, a membrane protein involved in signal transduction. Association of eNOS with caveolin is reversible, dissociation from caveolin activating the enzyme. Oxidised low-density lipoprotein (oxLDL) displaces eNOS from caveolae by binding to endothelial cell CD36 receptors. This depletes the caveolae of cholesterol, disturbing eNOS function.
The nitrogen atom in NO is derived from the terminal guanidino group of L-arginine. NOS enzymes are functionally ‘bimodal’, in that they combine oxygenase and reductase activities associated with distinct structural domains. The oxygenase domain contains haem, while the reductase domain binds calcium–calmodulin. In pathological states, the enzyme can undergo structural change leading to electron transfer between substrates, enzyme co-factors and products becoming ‘uncoupled’, so that electrons are transferred to molecular oxygen, leading to the synthesis of superoxide anion (O2−) rather than NO. This is important, as superoxide anion reacts with NO to form a toxic product (peroxynitrite anion; see p. 240 below).
L-Arginine is usually present in excess in endothelial cell cytoplasm, so the rate of production of NO is determined by the activity of the enzyme rather than by substrate availability. Nevertheless, very high doses of L-arginine can restore endothelial NO biosynthesis in some pathological states (e.g. hypercholesterolaemia; see below) in which endothelial function is impaired. Possible explanations for this paradox include:
•
compartmentation: i.e. existence of a distinct pool of substrate in a cell compartment with access to the synthase enzyme, which can become depleted despite apparently plentiful total cytoplasmic arginine concentrations
•
competition with endogenous inhibitors of NOS such as
asymmetric dimethylarginine (ADMA; see below), which is elevated in plasma from patients with hypercholesterolaemia
•
reassembly/reactivation of enzyme in which transfer of electrons has become uncoupled from
L-arginine as a result of an action of supraphysiological concentrations of
L-arginine.
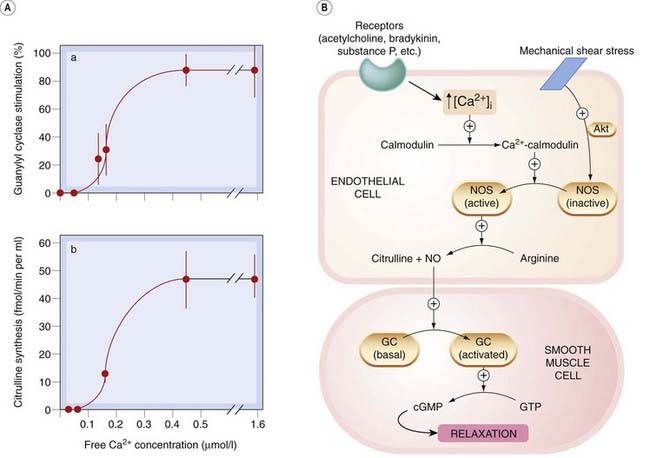
The activity of constitutive isoforms of NOS is controlled by intracellular calcium–calmodulin (Fig. 20.3). Control is exerted in two ways:
1
Many endothelium-dependent agonists (e.g. acetylcholine, bradykinin, substance P) increase the cytoplasmic concentration of calcium ions, [Ca
2+]
i; the consequent increase in calcium–calmodulin activates eNOS or nNOS.
2
Phosphorylation of specific residues on eNOS controls its sensitivity to calcium–calmodulin; this can alter NO synthesis in the absence of any change in [Ca
2+]
i.
One important physiological stimulus controlling endothelial NO synthesis in resistance vessels is believed to be shear stress. This is sensed by endothelial mechanoreceptors and transduced via a serine–threonine protein kinase called Akt. Agonists that increase cAMP in endothelial cells (e.g. β2-adrenoceptor agonists) also increase eNOS activity, but via protein kinase A-mediated phosphorylation,2 whereas protein kinase C reduces eNOS activity by phosphorylating residues in the calmodulin-binding domain, thereby reducing the binding of calmodulin. Insulin increases eNOS activity via tyrosine kinase activation (and also increases the expression of nNOS in diabetic mice).
In contrast to constitutive NOS isoforms, the activity of iNOS is effectively independent of [Ca2+]i, being fully activated even at the low values of [Ca2+]i present under resting conditions. The enzyme is induced by bacterial lipopolysaccharide and/or cytokines synthesised in response to lipopolysaccharide, notably interferon-γ, the antiviral effect of which can be explained by this action. Tumour necrosis factor-α and interleukin-1 do not alone induce iNOS, but they each synergise with interferon-γ in this regard (see Ch. 17). Induction of iNOS is inhibited by glucocorticoids and by several cytokines, including transforming growth factor-β. There are important species differences in the inducibility of iNOS, which is less readily induced in human than in mouse cells.
Nitric oxide synthesis, inactivation and carriage 
•
Nitric oxide (NO) is synthesised from
L-arginine and molecular O
2 by nitric oxide synthase (NOS).
•
NOS exists in three isoforms: inducible, and constitutive endothelial and neuronal forms (respectively, iNOS, eNOS and nNOS). NOSs are dimeric flavoproteins, contain tetrahydrobiopterin and have homology with cytochrome P450. The constitutive enzymes are activated by calcium–calmodulin. Sensitivity to calcium–calmodulin is controlled by phosphorylation of specific residues on the enzymes.
•
iNOS is induced in macrophages and other cells by interferon-γ.
•
nNOS is present in the central nervous system (see
Chs 36–
38) and in autonomic nerves (see
Ch. 12).
•
eNOS is present in platelets and other cells in addition to endothelium.
•
NO is inactivated by combination with the haem of haemoglobin or by oxidation to nitrite and nitrate, which are excreted in urine.
•
NO is unstable but can react reversibly with cysteine residues (e.g. in globin or albumin) to form stable nitrosothiols; as a result, red cells can act as an O
2-regulated source of NO. NO released in this way escapes inactivation by haem by being exported via cysteine residues in the anion exchange protein in red cell membranes.
Degradation and Carriage of Nitric Oxide
Nitric oxide reacts with oxygen to form N2O4, which combines with water to produce a mixture of nitric and nitrous acids. Nitrite ions are oxidised to nitrate by oxyhaemoglobin. These reactions are summarised as follow:
(20.1) 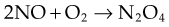
(20.2) 
(20.3) 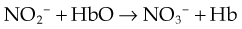
Low concentrations of NO are relatively stable in air, so small amounts of NO produced in the lung escape degradation and can be detected in exhaled air. In contrast, NO reacts very rapidly with even low concentrations of superoxide anion (O2−) to produce peroxynitrite anion (ONOO−), which is responsible for some of its toxic effects. Haem has an affinity for NO > 10 000 times greater than for oxygen. In the absence of oxygen, NO bound to haem is relatively stable, but in the presence of oxygen NO is converted to nitrate and the haem iron oxidised to methaemoglobin.
Endothelium-derived NO acts locally on underlying vascular smooth muscle or on adherent monocytes or platelets. A strong, but still controversial, case has been made that NO can also act at a distance in the mammalian circulation via reversible interactions with haemoglobin.3
Distinct from the inactivation reaction between NO and haem, a specific cysteine residue in globin combines reversibly with NO under physiological conditions. It is proposed that the resulting S-nitrosylated haemoglobin acts as a circulating oxygen-sensitive NO carrier. Albumin can also be reversibly nitrosylated and could function similarly, as could the inorganic nitrite ion—indeed, foods rich in inorganic nitrate (reduced to nitrite in vivo by anaerobic organisms in the mouth) have potential for prevention of vascular disease; see below. Readers who require a more detailed account of the case that NO acts at a distance within the mammalian circulation are directed to reviews by Singel & Stamler (2005) and, for a sceptical view, Schechter & Gladwyn (2003).
Effects of Nitric Oxide
Nitric oxide reacts with various metals, thiols and oxygen species, thereby modifying proteins, DNA and lipids. One of its most important biochemical effects (see Ch. 3) is activation of soluble guanylyl cyclase, a heterodimer present in vascular and nervous tissue as two distinct isoenzymes. Guanylyl cyclase synthesises the second messenger cGMP. NO activates the enzyme by combining with its haem group, and many physiological effects of low concentrations of NO are mediated by cGMP. These effects are prevented by inhibitors of guanylyl cyclase (e.g. 1H-[1,2,4]-oxadiazole-[4,3-α]-quinoxalin-1-one, better known as ‘ODQ’), which are useful investigational tools. NO activates soluble guanylyl cyclase in intact cells (neurons and platelets) extremely rapidly, and activation is followed by desensitisation to a steady-state level. This contrasts with its effect on the isolated enzyme, which is slower but more sustained. Guanylyl cyclase contains another regulatory site, which is NO independent. This is activated by several investigational drugs.
Effects of cGMP are terminated by phosphodiesterase enzymes. Sildenafil and tadalafil are inhibitors of phosphodiesterase type V that are used to treat erectile dysfunction, because they potentiate NO actions in the corpora cavernosa of the penis by this mechanism (see Ch. 34). NO also combines with haem groups in other biologically important proteins, notably cytochrome c oxidase, where it competes with oxygen, contributing to the control of cellular respiration (see Erusalimsky & Moncada, 2007). Cytotoxic and/or cytoprotective effects of higher concentrations of NO relate to its chemistry as a free radical (see Ch. 37). Some physiological and pathological effects of NO are shown in Table 20.1.
Biochemical and Cellular Aspects
Pharmacological effects of NO can be studied with NO gas dissolved in deoxygenated salt solution. More conveniently, but less directly, various donors of NO, such as nitroprusside, S-nitrosoacetylpenicillamine (SNAP) or S-nitrosoglutathione (SNOG), have been used as surrogates. This has pitfalls; for example, ascorbic acid potentiates SNAP but inhibits responses to authentic NO.4
Nitric oxide can activate guanylyl cyclase in the same cells that produce it, giving rise to autocrine effects, for example on the barrier function of the endothelium. NO also diffuses from its site of synthesis and activates guanylyl cyclase in neighbouring cells. The resulting increase in cGMP affects protein kinase G, cyclic nucleotide phosphodiesterases, ion channels and possibly other proteins. This inhibits the [Ca2+]i-induced smooth muscle contraction and platelet aggregation that occur in response to contractile or proaggregatory agonists. NO also hyperpolarises vascular smooth muscle, as a consequence of potassium channel activation. NO inhibits monocyte adhesion and migration, adhesion and aggregation of platelets, and smooth muscle and fibroblast proliferation. These cellular effects probably underlie the antiatherosclerotic action of NO (see Ch. 23).
Large amounts of NO (released following induction of NOS or excessive stimulation of NMDA receptors in the brain) cause cytotoxic effects (either directly or via peroxynitrite anions). These contribute to host defence, but also to the neuronal destruction that occurs when there is overstimulation of NMDA receptors by glutamate (see Chs 37 and 39). Paradoxically, NO is also cytoprotective under some circumstances (see Ch. 39).
Vascular Effects (See Also Ch. 22)
The L-arginine/NO pathway is tonically active in resistance vessels, reducing peripheral vascular resistance and hence systemic blood pressure. Mutant mice that lack the gene coding for eNOS are hypertensive, consistent with a role for NO biosynthesis in the physiological control of blood pressure. In addition, NO derived from nNOS has recently been implicated in the control of basal resistance vessel tone in human forearm and cardiac muscle vascular beds (Seddon et al., 2008, 2009). Increased NO generation may contribute to the generalised vasodilatation that occurs during pregnancy.
Neuronal Effects (See Also Ch. 12)
Nitric oxide is a non-noradrenergic non-cholinergic (NANC) neurotransmitter in many tissues (Ch. 12), and is important in the upper airways, gastrointestinal tract and control of penile erection (Chs 27, 29 and 34). It is implicated in the control of neuronal development and of synaptic plasticity in the CNS (Chs 36 and 38). Mice carrying a mutation disrupting the gene coding nNOS have grossly distended stomachs similar to those seen in human hypertrophic pyloric stenosis (a disorder characterised by pyloric hypertrophy causing gastric outflow obstruction, which occurs in approximately 1 in 150 male infants and is corrected surgically). nNOS knockout mice resist stroke damage caused by middle cerebral artery ligation but are aggressive and oversexed (characteristics that may not be unambiguously disadvantageous, at least in the context of natural selection!).
Host Defence (See Ch. 17)
Cytotoxic and/or cytostatic effects of NO are implicated in primitive non-specific host defence mechanisms against numerous pathogens, including viruses, bacteria, fungi, protozoa and parasites, and against tumour cells. The importance of this is evidenced by the susceptibility to Leishmania major (to which wild-type mice are highly resistant) of mice lacking iNOS. Mechanisms whereby NO damages invading pathogens include nitrosylation of nucleic acids and combination with haem-containing enzymes, including the mitochondrial enzymes involved in cell respiration.
Actions of nitric oxide
•
Nitric oxide (NO) acts by:
–
combining with haem in guanylyl cyclase, activating the enzyme, increasing cGMP and thereby lowering [Ca
2+]
i–
combining with haem groups in other proteins (e.g. cytochrome
c oxidase)
–
combining with superoxide anion to yield the cytotoxic peroxynitrite anion
–
nitrosation of proteins, lipids and nucleic acids.
•
Effects of NO include:
–
vasodilatation, inhibition of platelet and monocyte adhesion and aggregation, inhibition of smooth muscle proliferation, protection against atheroma
–
synaptic effects in the peripheral and central nervous system
–
host defence and cytotoxic effects on pathogens
Therapeutic Approaches
Nitric Oxide
Inhalation of high concentrations of NO (as occurred when cylinders of nitrous oxide, N2O, for anaesthesia were accidentally contaminated) causes acute pulmonary oedema and methaemoglobinaemia, but concentrations below 50 ppm (parts per million) are not toxic. NO (5–300 ppm) inhibits bronchoconstriction (at least in guinea pigs), but the main action of inhaled NO is pulmonary vasodilatation. Inspired NO acts preferentially on ventilated alveoli, and could therefore be therapeutically useful in respiratory distress syndrome. This condition has a high mortality and is caused by diverse insults (e.g. infection). It is characterised by intrapulmonary ‘shunting’ (i.e. pulmonary arterial blood entering the pulmonary vein without passing through capillaries in contact with ventilated alveoli), resulting in arterial hypoxaemia, and by acute pulmonary arterial hypertension. Inhaled NO dilates blood vessels in ventilated alveoli (which are exposed to the inspired gas) and thus reduces shunting. NO is used in intensive care units to reduce pulmonary hypertension and to improve oxygen delivery in patients with respiratory distress syndrome, but it is not known whether this improves long-term survival in these severely ill patients. Ethyl nitrite gas has been investigated in newborns (who are at much increased risk of respiratory distress syndrome because of their immature lungs) as a potentially less toxic alternative.
Nitric Oxide Donors/Precursors
Nitrovasodilators have been used therapeutically for over a century. The common mode of action of these drugs is as a source of NO (Chs 21 and 22). There is interest in the potential for selectivity of nitrovasodilators; for instance, glyceryl trinitrate is more potent on vascular smooth muscle than on platelets, whereas SNOG (see above) selectively inhibits platelet function. It was shown recently that dietary nitrate (contained in beetroot juice) acutely lowers arterial blood pressure in parallel with a rise in plasma nitrite concentration and improved endothelial and platelet function. Interruption of the enterosalivary conversion of nitrate to nitrite prevents the rise in plasma nitrite, blocks the fall in blood pressure and abolishes the inhibitory effect on platelet aggregation (Webb et al., 2008).5
Inhibition of Nitric Oxide Synthesis
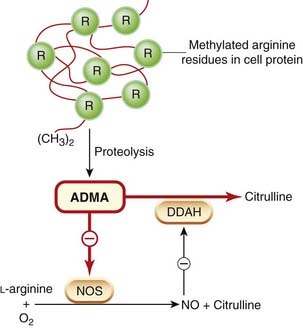
Drugs can inhibit NO synthesis or action by several mechanisms. Certain arginine analogues compete with arginine for NOS. Several such compounds, for example NG-monomethyl-L-arginine (L-NMMA) and NG-nitro-L-arginine methyl ester (L-NAME), have proved of great value as experimental tools. One such compound, ADMA (see above), is approximately equipotent with L-NMMA. It is present in human plasma and is excreted in urine. Its plasma concentration correlates with vascular mortality in patients receiving haemodialysis for chronic renal failure, and is increased in people with hypercholesterolaemia. In addition to urinary excretion, ADMA is also eliminated by metabolism to a mixture of citrulline and methylamine by dimethylarginine dimethylamino hydrolase (DDAH), an enzyme that exists in two isoforms, each with a functionally essential reactive cysteine residue in the active site that is subject to control by nitrosylation. Inhibition of DDAH by NO causes feedback inhibition of the L-arginine/NO pathway by allowing cytoplasmic accumulation of ADMA. Conversely, activation of DDAH could potentiate the L-arginine/NO pathway; see Figure 20.4.
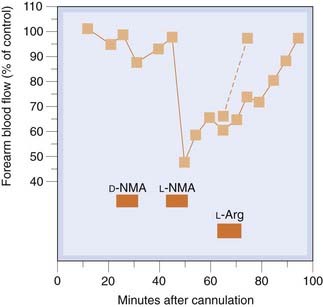
Infusion of a low dose of L-NMMA into the brachial artery causes local vasoconstriction (Fig. 20.5), owing to inhibition of the basal production of NO in the infused arm, probably by inhibiting nNOS (Seddon et al., 2008), without influencing blood pressure or causing other systemic effects, whereas intravenous L-NMMA causes vasoconstriction in renal, mesenteric, cerebral and striated muscle resistance vessels, increases blood pressure and causes reflex bradycardia.
There is therapeutic interest in selective inhibitors of different isoforms of NOS. Selective inhibitors of iNOS versus the two constitutive forms have been described (e.g. N-iminoethyl-L-lysine), and have potential for the treatment of inflammatory and other conditions in which iNOS has been implicated (e.g. asthma). 7-Nitroindazole selectively inhibits nNOS, the mechanism of selectivity being uncertain. S-methyl-L-thiocitrulline is a potent and selective inhibitor of human nNOS (Furfine et al., 1994), and has recently provided new understanding of the importance of nNOS in control of human resistance vessel tone in vivo (Seddon et al., 2008, 2009).
An endogenous protein inhibitor of nNOS (termed PIN) works by an entirely different mechanism, namely destabilising the NOS dimer (Jaffrey & Snyder, 1996).
Inhibition of the L-arginine/nitric oxide pathway
•
Glucocorticoids inhibit biosynthesis of inducible (but not constitutive) nitric oxide synthase (NOS).
•
Synthetic arginine analogues (e.g.
L-NMMA,
L-NAME; see text) compete with arginine and are useful experimental tools.
•
Endogenous NOS inhibitors include ADMA (see text) and PIN (a protein that inhibits NOS dimerisation).
•
Isoform-selective inhibitors include
S-methyl-
L-thiocitrulline (selective for nNOS).
Potentiation of Nitric Oxide
Several means whereby the L-arginine/NO pathway could be enhanced are under investigation. Some of these rely on existing drugs of proven value in other contexts. The hope (as yet unproven) is that, by potentiating NO, they will prevent atherosclerosis or its thrombotic complications or have other beneficial effects attributed to NO. Possibilities include:
•
selective NO donors as ‘replacement’ therapy (see above)
•
dietary supplementation with
L-arginine (see above)
•
antioxidants (to reduce concentrations of reactive oxygen species and hence stabilise NO;
Ch. 22)
•
drugs that restore endothelial function in patients with metabolic risk factors for vascular disease (e.g. angiotensin-converting enzyme inhibitors, statins, insulin, oestrogens;
Chs 22,
23,
30 and
34)
•
β
2-adrenoceptor agonists and related drugs (e.g.
nebivolol, a β
1-adrenoceptor antagonist that is metabolised to an active metabolite that activates the
L-arginine/NO pathway)
•
phosphodiesterase type V inhibitors (e.g.
sildenafil; see above and
Ch. 34).
Clinical Conditions in Which Nitric Oxide May Play A Part
The wide distribution of NOS enzymes and diverse actions of NO suggest that abnormalities in the L-arginine/NO pathway could be important in disease. Either increased or reduced production could play a part, and hypotheses abound. Evidence is harder to come by but has been sought using various indirect approaches, including:
•
analysing nitrate and/or cGMP in urine: these studies are bedevilled, respectively, by dietary nitrate and by membrane-bound guanylyl cyclase (which is stimulated by endogenous natriuretic peptides; see
Ch. 21)
•
a considerable refinement is to administer [
15N]-arginine and use mass spectrometry to measure the enrichment of
15N over naturally abundant [
14N]-nitrate in urine
•
measuring NO in exhaled air
•
measuring effects of NOS inhibitors (e.g.
L-NMMA)
•
comparing responses to endothelium-dependent agonists (e.g.
acetylcholine) and endothelium-independent agonists (e.g.
nitroprusside)
•
measuring responses to increased blood flow (‘flow-mediated dilatation’), which are largely mediated by NO
•
studying histochemical appearances and pharmacological responses in vitro of tissue obtained at operation (e.g. coronary artery surgery).
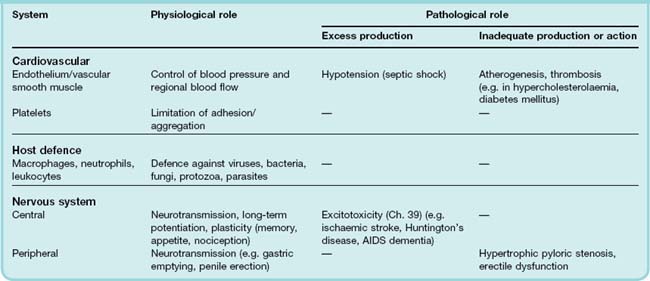
All these methods have limitations, and the dust is far from settled. Nevertheless, it seems clear that the L-arginine/NO pathway is indeed a player in the pathogenesis of several important diseases, opening the way to new therapeutic approaches. Some pathological roles of excessive or reduced NO production are summarised in Table 20.1. We touch only briefly on these clinical conditions, and would caution the reader that not all of these exciting possibilities are likely to withstand the test of time!
Nitric oxide in pathophysiology
•
Nitric oxide (NO) is synthesised under physiological and pathological circumstances.
•
Either reduced or increased NO production can contribute to disease.
•
Underproduction of neuronal NO is reported in babies with hypertrophic pyloric stenosis. Endothelial NO production is reduced in patients with hypercholesterolaemia and some other risk factors for atherosclerosis, and this may contribute to atherogenesis.
•
Overproduction of NO may be important in neurodegenerative diseases (see
Ch. 39) and in septic shock (
Ch. 22).
Sepsis can cause multiple organ failure. Whereas NO benefits host defence by killing invading organisms, excessive NO causes harmful hypotension. Disappointingly, however, L-NMMA worsened survival in one controlled clinical trial. Chronic low-grade endotoxaemia occurs in patients with hepatic cirrhosis. Systemic vasodilatation is typical in such patients. Urinary excretion of cGMP is increased, and vasodilatation may be a consequence of induction of NOS leading to increased NO synthesis. Nitrosative stress and nitration of proteins in airway epithelium may contribute to steroid resistance in asthma, and the ineffectiveness of glucocorticoids in chronic obstructive pulmonary disease (see Ch. 27).
Nitric oxide biosynthesis is reduced in patients with hypercholesterolaemia and some other disorders that predispose to atheromatous vascular disease, including cigarette smoking and diabetes mellitus. In hypercholesterolaemia, evidence of blunted NO release in forearm and coronary vascular beds is supported by evidence that this can be corrected by lowering plasma cholesterol (with a statin; see Ch. 24) or by dietary supplementation with L-arginine.
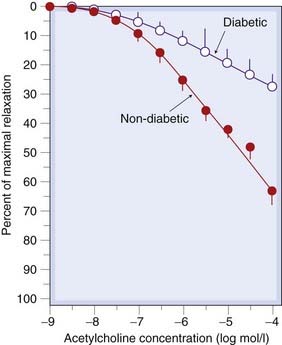
Endothelial dysfunction in diabetic patients with erectile dysfunction occurs in tissue from the corpora cavernosum of the penis, as evidenced by blunted relaxation to acetylcholine despite preserved responses to nitroprusside (Fig. 20.6). Vasoconstrictor responses to intra-arterial L-NMMA are reduced in forearm vasculature of insulin-dependent diabetics, especially in patients with traces of albumin in their urine (‘microalbuminuria’: early evidence of glomerular endothelial dysfunction), suggesting that basal NO synthesis may be reduced throughout their circulation.
It is thought that failure to increase endogenous NO biosynthesis normally during pregnancy contributes to eclampsia. This is a hypertensive disorder that accounts for many maternal deaths and in which the normal vasodilatation seen in healthy pregnancy is lost.
Excessive NMDA receptor activation increases NO synthesis, which contributes to several forms of neurological damage (see Ch. 39). nNOS is absent in pyloric tissue from babies with idiopathic hypertrophic pyloric stenosis.
Established clinical uses of drugs that influence the L-arginine/NO system are summarised in the clinical box.
Nitric oxide in therapeutics 
•
Nitric oxide (NO) donors (e.g.
nitroprusside and
organic nitrovasodilators) are well established (see
Chs. 21 and
22).
•
Type V phosphodiesterase inhibitors (e.g.
sildenafil,
tadalafil) potentiate the action of NO. They are used to treat erectile dysfunction (
Ch. 34).
•
Other possible uses (e.g. pulmonary hypertension, gastric stasis) are being investigated.
•
Inhaled NO is used in adult and neonatal respiratory distress syndrome.
•
Inhibition of NO biosynthesis is being investigated in disorders where there is overproduction of NO (e.g. inflammation and neurodegenerative disease). Disappointingly,
L-NMMA increases mortality in one such condition (sepsis).
References and Further Reading
Biochemical aspects
Bellamy T.C., Wood J., Goodwin D.A., Garthwaite J. Rapid desensitization of the nitric oxide receptor, soluble guanylyl cyclase, underlies diversity of cellular cGMP responses. Proc. Natl. Acad. Sci. USA.. 2000;97:2928-2933. In its natural environment, soluble guanylyl cyclase behaves much more like a neurotransmitter receptor than had been expected from previous enzymological studies; rapid desensitisation is likely to be important under physiological conditions
Fulton D., Fontana J., Sowa G., et al. Localization of endothelial nitric-oxide synthase phosphorylated on serine 1179 and nitric oxide in Golgi and plasma membrane defines the existence of two pools of active enzyme. J. Biol. Chem.. 2002;277:4277-4284. Activated, phosphorylated eNOS resides in caveolin-enriched plasmalemma and Golgi membranes; both pools are VEGF-regulated to produce NO
Furfine E.S., Harmon M.F., Paith J.E., et al. Potent and selective inhibition of human nitric oxide synthases: selective inhibition of neuronal nitric oxide synthase by S-methyl-L-thiocitrulline and S-ethyl-L-thiocitrulline. J. Biol. Chem.. 1994;269:26677-26683.
Hess D.T., Matsumoto A., Kim S.O., et al. Protein S-nitrosylation: purview and parameters. Nature Rev. Mol. Cell Biol.. 2005;6:150-166.
Jaffrey S.R., Snyder S. PIN: an associated protein inhibitor of neuronal nitric oxide synthase. Science. 1996;274:774-777. Works by destabilising the nNOS dimer
Kim-Shapiro D.B., Schechter A.N., Gladwin M.T. Unraveling the reactions of nitric oxide, nitrite, and hemoglobin in physiology and therapeutics. Arterioscler. Thromb. Vasc. Biol.. 2006;26:697-705. Reviews recent evidence that nitrite anion may be the main intravascular NO storage molecule; cf. Singel & Stamler, 2005, below
Krumenacker J., Hanafy K.A., Murad F. Regulation of nitric oxide and soluble guanylyl cyclase. Brain Res. Bull.. 2004;62:505-515.
Lee J., Ryu H., Ferrante R.J., et al. Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proc. Natl. Acad. Sci. USA. 2003;100:4843-4848. Inhibition of NOS activity by arginine depletion in stimulated astrocyte cultures occurs via inhibition of translation of iNOS mRNA, and provides one explanation for the ‘arginine paradox’ while indicating a distinct mechanism by which substrate can regulate the activity of its associated enzyme
Liu J., Garcia-Cardena G., Sessa W.C. Palmitoylation of endothelial nitric oxide synthase is necessary for optimal stimulated release of nitric oxide: implications for caveolae localization. Biochemistry. 1996;35:13277-13281. N-Myristoylation of eNOS is necessary for its association and targeting into the Golgi complex, whereas palmitoylation influences its targeting to caveolae
Matsubara M., Hayashi N., Jing T., Titani K. Regulation of endothelial nitric oxide synthase by protein kinase C. J. Biochem.. 2003;133:773-781. Protein kinase C inhibits eNOS activity by changing the binding of calmodulin to the enzyme
Pawloski J.R., Hess D.T., Stamler J.S. Export by red cells of nitric oxide bioactivity. Nature. 2001;409:622-626. Movement of NO from red blood cells via anion exchange protein AE1; see also editorial by Gross, S.S. pp. 577–578
Ribiero J.M.C., Hazzard J.M.H., Nussenzveig R.H., et al. Reversible binding of nitric oxide by a salivary haem protein from a blood sucking insect. Science. 1993;260:539-541. Action at a distance
Schechter A.N., Gladwyn M.T. Hemoglobin and the paracrine and endocrine functions of nitric oxide. N. Engl. J. Med.. 2003;348:1483-1485. See also dissenting correspondence in New Engl. J Med. 394, 402–406
Shaul P.W. Regulation of endothelial nitric oxide synthase: location, location, location. Annu. Rev. Physiol.. 2002;64:749-774.
Singel D.J., Stamler J.S. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu. Rev. Physiol.. 2005;67:99-145.
Stuehr D.J., Santolini J., Wang Z.Q., et al. Update on mechanism and catalytic regulation in the NO synthases. J. Biol. Chem.. 2004;279:36167-36170.
Xu W.M., Charles I.G., Moncada S. Nitric oxide: orchestrating hypoxia regulation through mitochondrial respiration and the endoplasmic reticulum stress response. Cell Res.. 2005;15:63-65.
Physiological aspects
Chiavegatto S., Nelson R.J. Interaction of nitric oxide and serotonin in aggressive behavior. Horm. Behav.. 2003;44:233-241. ‘NO appears to play an important role in normal brain 5-HT function and may have significant implications for the treatment of psychiatric disorders characterised by aggressive and impulsive behaviors.’
Coggins M.P., Bloch K.D. Nitric oxide in the pulmonary vasculature. Arterioscler. Thromb. Vasc. Biol.. 2007;27:1877-1885.
Diesen D.L., Hess D.T., Stamler J.S. Hypoxic vasodilation by red blood cells evidence for an S-nitrosothiol-based signal Circ. Res. 103:2008:545-553 Untreated human red blood cells [RBCs] relax thoracic aorta at low PO2 but not at high PO2. Relaxation is largely endothelium independent and unaffected by NO synthase inhibition or nitrite. Robust relaxations by RBCs are elicited in the absence of endothelial, neuronal or inducible NO synthase. The results suggest that an S-nitrosothiol originating from RBCs mediates hypoxic vasodilatation by RBCs
Erusalimsky J.D., Moncada S. Nitric oxide and mitochondrial signaling from physiology to pathophysiology Arterioscler. Thromb. Vasc. Biol. 27:2007:2524-2531 Reviews the evidence that binding of NO to cytochrome c oxidase elicits intracellular signalling events
Furchgott R.F., Zawadzki J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373-376. Classic
Nelson R.J., Demas G.E., Huang P.L., et al. Behavioural abnormalities in male mice lacking neuronal nitric oxide synthase. Nature. 1995;378:383-386. ‘A large increase in aggressive behaviour and excess, inappropriate sexual behaviour in nNOS knockout mice’
Seddon M.D., Chowienczyk P.J., Brett S.E., et al. Neuronal nitric oxide synthase regulates basal microvascular tone in humans in vivo. Circulation. 2008;117:1991-1996. Paradigm shift?—very possibly; see next reference
Seddon M., Melikian N., Dworakowski R., et al. Effects of neuronal nitric oxide synthase on human coronary artery diameter and blood flow in vivo. Circulation. 2009;119:2656-2662. Local nNOS-derived NO regulates basal blood flow in the human coronary vascular bed, whereas substance P-stimulated vasodilatation is eNOS mediated
Toda N., Okamura T. The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol. Rev.. 2003;55:271-324.
Vallance P., Leiper J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol.. 2004;24:1023-1030.
Victor V.M., Núñez C., D’Ocón P., et al Regulation of oxygen distribution in tissues by endothelial nitric oxide Circ. Res. 104:2009:1178-1183 Endogenously released endothelial NO inhibits cytochrome c oxidase and can modulate tissue O2 consumption and regulates O2 distribution to the surrounding tissues
Pathological aspects
Boger R.H., Tsikas D., Bode-Boger S.M., et al. Hypercholesterolemia impairs basal nitric oxide synthase turnover rate: a study investigating the conversion of L-[guanidino-15N2]-arginine to N-15-labeled nitrate by gas chromatography-mass spectrometry. Nitric Oxide Biol. Chem.. 2004;11:1-8. The mechanism of impaired NOS activity in hypercholesterolaemia most likely involves inhibition of NOS by ADMA
Ricciardolo F.L.M., Sterk P.J., Gaston B., et al. Nitric oxide in health and disease of the respiratory system. Physiol. Rev.. 2004;84:731-765.
Shaul P.W. Endothelial nitric oxide synthase, caveolae and the development of atherosclerosis. J. Physiol. (Lond). 2003;547:21-33. oxLDL displaces eNOS from caveolae by binding to endothelial cell CD36 receptors and by depleting caveolae cholesterol content, resulting in the disruption of eNOS activation; the adverse effects of oxLDL are prevented by high-density lipoprotein and could be involved in early phases of atherogenesis
Clinical and therapeutic aspects
Griffiths M.J.D., Evans T.W. Drug therapy: inhaled nitric oxide therapy in adults. N. Engl. J. Med.. 2005;353:2683-2695. Concludes that, on the available evidence, inhaled NO is not effective in patients with acute lung injury, but that it may be useful as a short-term measure in acute hypoxia ± pulmonary hypertension
Malmström R.E., Törnberg D.C., Settergren G., et al. Endogenous nitric oxide release by vasoactive drugs monitored in exhaled air. Am. J. Respir. Crit. Care Med.. 2003;168:114-120. In humans, acetylcholine evokes a dose-dependent increase of NO in exhaled air; NO release by vasoactive agonists can be measured online in the exhaled air of pigs and humans
Miller M.R., Megson I.L. Review—Recent developments in nitric oxide donor drugs. Br. J. Pharmacol.. 2007;151:305-321. Explores some of the more promising recent advances in NO donor drug development and challenges associated with NO as a therapeutic agent
Moya M.P., Gow A.J., Califf R.M., et al. Inhaled ethyl nitrite gas for persistent pulmonary hypertension of the newborn. Lancet. 2002;360:141-143. ‘Ethyl nitrite can improve oxygenation and systemic haemodynamics in neonates, and seems to reduce rebound hypoxaemia and production of toxic byproducts’
Pawloski J.R., Hess D.T., Stamler J.S. Impaired vasodilation by red blood cells in sickle cell disease Proc. Natl. Acad. Sci. USA 102:2005:2531-2536 Sickle red cells are deficient in membrane S-nitrosothiol and impaired in their ability to mediate hypoxic vasodilation; the magnitudes of these impairments correlate with the clinical severity of disease
Webb A.J., Patel N., Loukogeorgakis S., et al. Acute blood pressure lowering, vasoprotective, and antiplatelet properties of dietary nitrate via bioconversion to nitrite. Hypertension. 2008;51:784-790.