45 Antipsychotic drugs
Overview
In this chapter, we focus on schizophrenia and the drugs used to treat it. We start by describing the illness and what is known of its pathogenesis, including the various neurochemical hypotheses and their relation to the actions of the main types of antipsychotic drugs that are in use or in development.
Introduction
Psychotic illnesses include various disorders, but the term antipsychotic drugs—previously known as neuroleptic drugs, antischizophrenic drugs or major tranquillisers—conventionally refers to those used to treat schizophrenia, one of the most common and debilitating forms of mental illness. These same drugs are also used to treat mania (Ch. 46) and other acute behavioural disturbances (see clinical box on p. 562). Pharmacologically, most are dopamine receptor antagonists, although many of them also act on other targets, particularly 5-hydroxytryptamine (5-HT) receptors, which may contribute to their clinical efficacy. Existing drugs have many drawbacks in terms of their efficacy and side effects. Gradual improvements have been achieved with the newer drugs, but radical new approaches will probably have to wait until we have a better understanding of the causes and underlying pathology of the disease, which are still poorly understood.1
The Nature of Schizophrenia
Schizophrenia2 (see Stahl, 2008) affects about 1% of the population. It is one of the most important forms of psychiatric illness, because it affects young people, is often chronic and is usually highly disabling. There is a strong hereditary factor in its aetiology, and evidence suggestive of a fundamental biological disorder (see below). The main clinical features of the disease are as follow.
Positive symptoms
Negative symptoms
In addition, deficits in cognitive function (e.g. attention, memory) are often present,3 together with anxiety, guilt, depression and self punishment, leading to suicide attempts in up to 50% of cases, about 10% of which are successful. The clinical phenotype varies greatly, particularly with respect to the balance between positive and negative symptoms, and this may have a bearing on the efficacy of antipsychotic drugs in individual cases. Schizophrenia can present dramatically, usually in young people, with predominantly positive features such as hallucinations, delusions and uncontrollable behaviour, or more insidiously in older patients with negative features such as flat mood and social withdrawal. The latter may be more debilitated than those with a florid presentation, and the prognosis is generally worse. Schizophrenia can follow a relapsing and remitting course, or be chronic and progressive, particularly in cases with a later onset. Chronic schizophrenia used to account for most of the patients in long-stay psychiatric hospitals; following the closure of many of these in the UK, it now accounts for many of society’s outcasts.
A characteristic feature of schizophrenia is a defect in ‘selective attention’. Whereas a normal individual quickly accommodates to stimuli of a familiar or inconsequential nature, and responds only to stimuli that are unexpected or significant, the ability of schizophrenic patients to discriminate between significant and insignificant stimuli seems to be impaired. Thus, the ticking of a clock may command as much attention as the words of a companion; a chance thought, which a normal person would dismiss as inconsequential, may become an irresistible imperative.
Aetiology and Pathogenesis of Schizophrenia
Genetic and Environmental Factors
The cause of schizophrenia remains unclear but involves a combination of genetic and environmental factors (see Stahl, 2008). Thus a person may have a genetic makeup, probably an abnormality in more than just a single gene, that predisposes them to schizophrenia, but exposure to environmental factors may be required for schizophrenia to develop.
The disease shows a strong, but incomplete, hereditary tendency. In first-degree relatives, the risk is about 10%, but even in monozygotic (identical) twins, one of whom has schizophrenia, the probability of the other being affected is only about 50%, pointing towards the importance of environmental factors. Genetic linkage studies have identified more than 20 potential susceptibility genes (see Craddock et al., 2005; Harrison & Weinberger, 2005), but it is clear that no single gene is responsible. There are significant associations between polymorphisms in individual genes and the likelihood of an individual developing schizophrenia, but many are quite weak, and there appears to be no single gene that has an overriding influence.
 The most robust associations are with genes that control neuronal development, synaptic connectivity and glutamatergic neurotransmission. These include neuregulin, dysbindin and DISC-1. Transgenic mice that underexpress neuregulin-1, a protein involved in synaptic development and plasticity and which controls NMDA receptor expression, show a phenotype resembling human schizophrenia in certain respects. Malfunction of NMDA receptors is further implicated by genetic association with the genes for D-amino acid oxidase (DAAO), the enzyme responsible for making D-serine, an allosteric modulator of NMDA receptors (see Ch. 37), and for DAAO activator (G72). Dysbindin is located in postsynaptic density domains and may be involved in tethering receptors including NMDA receptors. DISC-1—which stands for disrupted in schizophrenia-1—is a protein that associates with cytoskeletal proteins and is involved with cell migration, neurite outgrowth and receptor trafficking. Among the other suggested susceptibility genes, some (such as the genes for monoamine oxidase A [MAO-A], tyrosine hydroxylase and the D2 dopamine receptor) are involved in monoamine transmission in the CNS. However, the weight of current evidence seems to suggest that schizophrenia may result from abnormal glutamatergic transmission involving a decrease in NMDA receptor function (see below).
The most robust associations are with genes that control neuronal development, synaptic connectivity and glutamatergic neurotransmission. These include neuregulin, dysbindin and DISC-1. Transgenic mice that underexpress neuregulin-1, a protein involved in synaptic development and plasticity and which controls NMDA receptor expression, show a phenotype resembling human schizophrenia in certain respects. Malfunction of NMDA receptors is further implicated by genetic association with the genes for D-amino acid oxidase (DAAO), the enzyme responsible for making D-serine, an allosteric modulator of NMDA receptors (see Ch. 37), and for DAAO activator (G72). Dysbindin is located in postsynaptic density domains and may be involved in tethering receptors including NMDA receptors. DISC-1—which stands for disrupted in schizophrenia-1—is a protein that associates with cytoskeletal proteins and is involved with cell migration, neurite outgrowth and receptor trafficking. Among the other suggested susceptibility genes, some (such as the genes for monoamine oxidase A [MAO-A], tyrosine hydroxylase and the D2 dopamine receptor) are involved in monoamine transmission in the CNS. However, the weight of current evidence seems to suggest that schizophrenia may result from abnormal glutamatergic transmission involving a decrease in NMDA receptor function (see below).
Some environmental influences early in development have been identified as possible predisposing factors, particularly maternal virus infections. This and other evidence suggests that schizophrenia is associated with a neurodevelopmental disorder affecting mainly the cerebral cortex and occurring in the first few months of prenatal development (see Harrison, 1997). This view is supported by brain-imaging studies showing cortical atrophy apparent in the early course of the disease which may increase with time and correlate with the progression of the disorder (van Haren et al., 2007). Studies of postmortem schizophrenic brains show evidence of misplaced cortical neurons with abnormal morphology. Other environmental factors such as cannabis consumption in adolescence and early adulthood (see Ch. 18) may also reveal schizophrenia.
The Neuroanatomical and Neurochemical Basis of Schizophrenia
Different symptoms of schizophrenia appear to result from malfunctions in different neuronal circuits. Changes in the mesolimbic pathway (the neuronal projection from the ventral tegmental area (VTA) to the nucleus accumbens, amygdala and hippocampus) being associated with positive symptoms, whereas negative and cognitive impairment symptoms are associated with changes in the mesocortical pathway (the projection from the VTA to areas of the prefrontal cortex).
The main neurotransmitters involved in the pathogenesis of schizophrenia are dopamine and glutamate.
Dopamine
The original dopamine theory of schizophrenia was proposed by Carlson—awarded a Nobel Prize in 2000—on the basis of indirect pharmacological evidence in humans and experimental animals. Amphetamine releases dopamine in the brain and can produce in humans a behavioural syndrome indistinguishable from an acute schizophrenic episode—very familiar to doctors who treat drug users. Also, hallucinations are a side effect of L-dopa therapy for Parkinson’s disease (see Ch. 39). In animals, dopamine release causes a specific pattern of stereotyped behaviour that resembles the repetitive behaviours sometimes seen in schizophrenic patients. Potent D2 receptor agonists, such as bromocriptine, produce similar effects in animals, and these drugs, like amphetamine, exacerbate the symptoms of schizophrenic patients. Furthermore, dopamine antagonists and drugs that block neuronal dopamine storage (e.g. reserpine) are effective in controlling the positive symptoms of schizophrenia, and in preventing amphetamine-induced behavioural changes.
It is now realised that the role of dopamine in schizophrenia is quite complex in that positive symptoms are thought to result from overactivity in the mesolimbic dopaminergic pathway activating D2 receptors (for a more detailed description of the dopamine pathways in the brain, see Ch. 38) whereas negative symptoms may result from a decreased activity in the mesocortical dopaminergic pathway where D1 receptors predominate (see Toda & Abi-Dargham, 2007). Other dopaminergic pathways in the brain (i.e. nigrostriatal and tuberoinfundibular; see Ch. 38) appear to function normally in schizophrenics.
There is a strong correlation between antipsychotic potency in reducing positive symptoms and activity in blocking D2 receptors (Fig. 45.1) and receptor-imaging studies have shown that clinical efficacy of antipsychotic drugs is consistently achieved when D2 receptor occupancy reaches about 80%.4 Furthermore, brain imaging studies have revealed an increased dopamine release in the striatum of schizophrenic patients (Laruelle et al., 1999).5 Injection of amphetamine caused dopamine release that was greater by a factor of two or more in schizophrenic subjects compared with control subjects, implying a greater amphetamine-induced release of dopamine. The effect was greatest in schizophrenic individuals during acute attacks, and absent during spontaneous remissions—clear evidence linking dopamine release to the symptomatology.
Thus, therapeutically it might be desirable to inhibit dopaminergic transmission in the limbic system yet enhance dopaminergic transmission in the prefrontal cortex (see below how this might be achieved).
Glutamate
In humans, NMDA receptor antagonists such as phencyclidine, ketamine and dizocilpine (Ch. 37) can produce both positive and negative psychotic symptoms—in contrast to amphetamine which produces only positive symptoms. It has therefore been postulated that schizophrenia may result from disruption of glutamatergic neurotransmission (Moghaddam, 2003), evident as a reduction in the function of NMDA receptors (the NMDA hypofunction hypothesis; see Coyle, 2006). Although schizophrenia is difficult to diagnose in a mouse, transgenic mice in which NMDA receptor expression is reduced (not abolished, because this is fatal) show stereotypic behaviours and reduced social interaction that are suggestive of schizophrenia and that respond to antipsychotic drugs.
Glutamatergic neurons and GABAergic neurons play complex roles in controlling the level of neuronal activity in both the mesocortical and the mesolimbic dopaminergic pathways. NMDA receptor hypofunction is thought to reduce the level of activity in mesocortical dopaminergic neurons. This would result in a decrease in dopamine release in the prefrontal cortex and thus give rise to negative symptoms of schizophrenia. On the other hand, NMDA receptor hypofunction is thought to enhance activity in the mesolimbic dopaminergic pathway, perhaps because in this pathway the important NMDA receptors are those located on GABAergic interneurons. Thus NMDA receptor hypofunction would result in reduced GABAergic inhibition (disinhibition) of mesolimbic dopaminergic neurons and thus give rise to enhanced dopamine release in limbic areas such as the nucleus accumbens, resulting in the production of positive symptoms
Given the evidence that schizophrenic symptoms may be due to a reduction in NMDA receptor function, efforts have been made to develop new drugs to enhance NMDA receptor function but not to a level where it becomes neurotoxic (see Ch. 39). This could be achieved by activating the facilitatory glycine site on the NMDA receptor (see Ch. 37) with an agonist (Shim et al., 2008) or by raising extracellular glycine levels by inhibiting the GlyT1 transporter (Bridges et al., 2008). AMPAkines, agents that allosterically enhance the action of glutamate at the AMPA receptor, by enhancing glutamate-induced neuronal depolarisation, can potentiate NMDA responses. Paradoxically, reducing glutamate release by activating presynaptic mGluR2/3 autoreceptors may result in a compensatory upregulation of NMDA receptors which also might be beneficial. This provides a novel target for the development of new antipsychotic drugs (see below).
Other glutamate pathways thought to be involved in schizophrenia are the corticostriatal, thalamocortical, corticothalamic and cortico-brain stem pathways. The thalamus normally functions as a sensory filter to limit unnecessary sensory input to the cortex. Disruption of the normal inputs to the thalamus, for example from a reduction in glutamatergic or GABAergic transmission, disables this ‘sensory gate’ function, allowing uninhibited input to reach the cortex. The role of the thalamus in schizophrenia is reviewed by Sim et al. (2006).
Neurodegeneration
Factors such as structural abnormalities in the brains of schizophrenics and progression of the disease—absence of symptoms in early childhood, the likelihood of positive symptoms becoming apparent before negative symptoms, progressive worsening, reduced responsiveness to drugs with time and the development of dementia—all indicate the involvement of ongoing neurodegeneration in the disease. The causes of such neurodegeneration are unclear at present but may involve glutamate-induced excitotoxicity (see Ch. 39).
The hope is that a fuller understanding of the altered function of glutamate transmission in schizophrenia will lead to the next generation of antipsychotic drugs (see Javitt, 2004).
The nature of schizophrenia 
Antipsychotic Drugs
Classification of Antipsychotic Drugs
More than 30 different antipsychotic drugs are available for clinical use. These can be divided into two groups—those drugs that were originally developed (e.g. chlorpromazine, haloperidol and many similar compounds), often referred to as first-generation, typical or conventional antipsychotic drugs, and more recently developed agents (e.g. clozapine, risperidone), which are termed atypical antipsychotic drugs. The term ‘atypical’ is widely used but not clearly defined (see Remington, 2003). In effect, it refers to the diminished tendency of the newer compounds to cause unwanted motor side effects (see below), but it is also used to describe compounds with a different pharmacological profile from first-generation compounds; several of these newer compounds improve the negative as well as the positive symptoms. In practice, however, it often serves—not very usefully—to distinguish the large group of similar first-generation dopamine antagonists from the more diverse group of newer compounds described below.
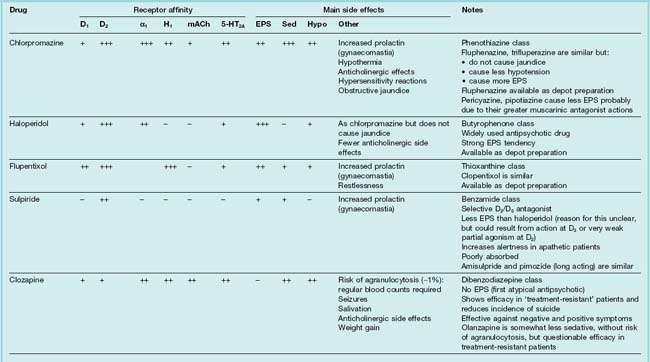
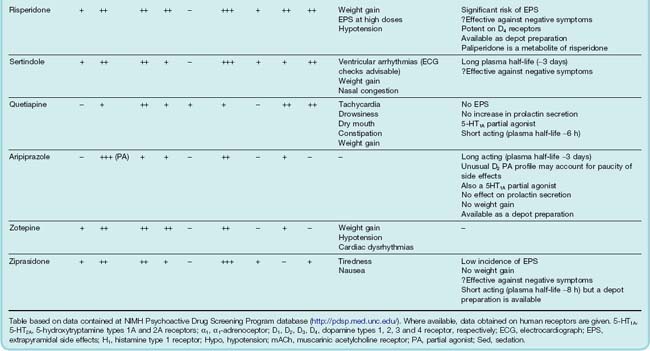
Table 45.1 summarises the main drugs that are in clinical use.
The therapeutic activity of the prototype drug, chlorpromazine, in schizophrenic patients was discovered through the acute observations of a French surgeon, Laborit, in 1947. He tested various substances, including promethazine, for their ability to alleviate signs of stress in patients undergoing surgery, and concluded that promethazine had a calming effect that was different from mere sedation. Elaboration of the phenothiazine structure led to chlorpromazine, the antipsychotic effect of which was demonstrated, at Laborit’s instigation, by Delay and Deniker in 1953. This drug was unique in controlling the symptoms of psychotic patients without excessively sedating them. The clinical efficacy of phenothiazines was discovered long before their mechanism was guessed at (let alone understood).
Pharmacological investigation showed that phenothiazines, the first-generation antipsychotic agents, block many different mediators, including histamine, catecholamines, acetylcholine and 5-HT, and this multiplicity of actions led to the trade name Largactil for chlorpromazine. It is now clear (see Fig. 45.1) that antagonism of dopamine is the main determinant of antipsychotic action.
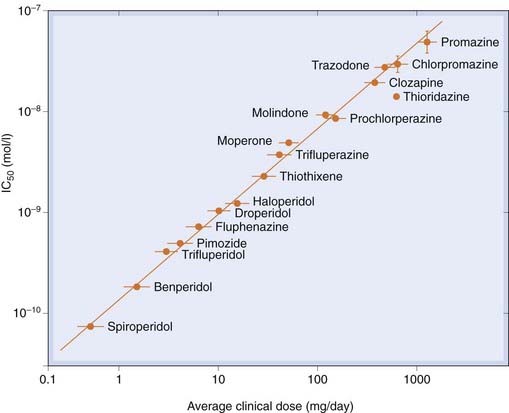
Fig. 45.1 Correlation between the clinical potency and affinity for dopamine D2 receptors among antipsychotic drugs.
Clinical potency is expressed as the daily dose used in treating schizophrenia, and binding activity is expressed as the concentration needed to produce 50% inhibition of haloperidol binding.
(From Seeman P et al. 1976 Nature 361: 717.)
Pharmacological Properties
Dopamine Receptors
The classification of dopamine receptors in the central nervous system is discussed in Chapter 38 (see Table 38.1). There are five subtypes, which fall into two functional classes: the D1 type, comprising D1 and D5, and the D2 type, comprising D2, D3 and D4. Antipsychotic drugs owe their therapeutic effects mainly to blockade of D2 receptors.6 As stated above, antipsychotic effects require about 80% block of D2 receptors. The first-generation compounds show some preference for D2 over D1 receptors, whereas some of the newer agents (e.g. sulpiride, amisulpride, remoxipride) are highly selective for D2 receptors. More recently, D2 antagonists that dissociate rapidly from the receptor and D2 partial agonists have been introduced in an attempt to reduce extrapyramidal motor side effects (see below).
It is the antagonism of D2 receptors in the mesolimbic pathway that is believed to relieve the positive symptoms of schizophrenia. Unfortunately, systemically administered antipsychotic drugs do not discriminate between D2 receptors in distinct brain regions and D2 receptors in other brain pathways will also be blocked. Thus, antipsychotic drugs produce unwanted motor effects (block of D2 receptors in the nigrostriatal pathway), enhance prolactin secretion (block of D2 receptors in the tuberoinfundibular pathway), reduce pleasure (block of D2 receptors in the reward component of the mesolimbic pathway) and perhaps even worsen the negative symptoms of schizophrenia (block of D2 receptors in the prefrontal cortex, although these are only expressed at a low density—D1 receptors being in greater abundance). While all antipsychotic drugs block D2 receptors and should therefore in theory induce all of these unwanted effects, some have additional pharmacological activity (e.g. mACh receptor antagonism and 5-HT2A receptor antagonism) that, to varying degrees, ameliorate unwanted effects (see below). 5-HT2A antagonism may help to alleviate the negative and cognitive impairments of schizophrenia.
Antipsychotic drugs have classically been thought to have a delayed onset to their therapeutic actions, even though their dopamine receptor-blocking action is immediate. This view has, however, been called into question (Kapur et al., 2005; Leucht et al., 2005). In animal studies, chronic antipsychotic drug administration does produce compensatory changes in the brain, for example a reduction in the activity of dopaminergic neurons and proliferation of dopamine receptors, detectable as an increase in haloperidol binding (see Seeman, 1987), with a pharmacological supersensitivity to dopamine reminiscent of the phenomenon of denervation supersensitivity (Ch. 12). The mechanism(s) of these delayed effects are poorly understood. They are likely to contribute to the development of unwanted tardive dyskinesias (see below). The sedating effect of antipsychotic drugs occurs extremely rapidly, allowing them to be used in acute behavioural emergencies.
Mechanism of action of antipsychotic drugs
5-Hydroxytryptamine Receptors
The idea that 5-HT dysfunction could be involved in schizophrenia has drifted in and out of favour many times (see Busatto & Kerwin, 1997). It was originally based on the fact that LSD, a partial agonist at 5-HT2A receptors (see Chs 15 & 47) produces hallucinations. Nowadays, conventional wisdom is that 5-HT may not be directly involved in the pathogenesis of schizophrenia. Nevertheless, pharmacological manipulation of 5-HT receptor activity, combined with D2 receptor antagonism, has resulted in new drugs with improved therapeutic profiles.7 There is a plethora of 5-HT receptors (see Chs 15 & 38) with disparate functions in the body (see also Chs 46 and 47). It is the 5-HT2A receptor and, to a lesser extent, the 5-HT1A receptor that are important in the treatment of schizophrenia.
5-HT2A receptors are Gi/Go-coupled receptors and their activation produces neuronal inhibition (through decreased neuronal excitability at the soma and decreased transmitter release at the nerve terminals; see Ch. 38). In this way, in the nigrostriatal pathway, 5-HT2A receptors control the release of dopamine. Drugs with 5-HT2A antagonist properties (e.g. olanzapine and risperidone) enhance dopamine release in the striatum by reducing the inhibitory effect of 5-HT. This will reduce extrapyramidal side effects (see below). In contrast, in the mesolimbic pathway, the combined effects of D2 and 5-HT2A antagonism are thought to counteract the increased dopamine function that gives rise to positive symptoms of schizophrenia. Further, enhancing both dopamine and glutamate release in the mesocortical circuit, 5-HT2A receptor antagonism may improve the negative symptoms of schizophrenia (Stahl, 2008).
5-HT1A receptors are somatodendritic autoreceptors that inhibit 5-HT release (see Ch. 38). Antipsychotic drugs that are agonists or partial agonists at 5-HT1A receptors (e.g. quetiapine; see Table 45.1) may work by decreasing 5-HT release thus enhancing dopamine release in the striatum and prefrontal cortex.
Muscarinic Acetylcholine Receptors
Some phenothiazine antipsychotic drugs (e.g. pericyazine) induce fewer extrapyramidal side effects than others, and this correlates with their affinity as muscarinic antagonists. Also, some newer, atypical drugs possess muscarinic antagonist properties (e.g. olanzepine). In the striatum, dopaminergic nerve terminals are thought to innervate cholinergic interneurons that express inhibitory D2 receptors (Pisani et al., 2007). It is suggested that there is normally a balance between D2 receptor activation and muscarinic receptor activation. Blocking D2 receptors in the striatum with an antipsychotic agent will result in enhanced acetylcholine release on to muscarinic receptors, thus producing extrapyramidal side effects, which are counteracted if the D2 antagonist also has muscarinic antagonist activity. Maintaining the dopamine/acetylcholine balance was also the rationale for the use of benztropine to reduce extrapyramidal effects of antipsychotic drugs (see Ch. 39). Muscarinic antagonist activity does, however, induce side effects such as constipation, dry mouth and blurred vision.
Behavioural Effects
Antipsychotic drugs produce many behavioural effects in experimental animals (see Ögren, 1996), but no single test distinguishes them clearly from other types of psychotropic drug. There are no good animal models of schizophrenia. For this reason, some pharmaceutical companies have even considered bypassing animal models, taking novel compounds directly from in vitro receptor assays to toxicology and preliminary clinical trials.
Antipsychotic drugs reduce spontaneous motor activity and in larger doses cause catalepsy, a state in which the animal remains immobile even when placed in an unnatural position. Inhibition of the hyperactivity induced by amphetamine parallels antipsychotic actions of these drugs, whereas their tendency to induce catalepsy parallels extrapyramidal symptoms (see below). Other tests reveal effects distinct from motor inhibition. For example, animals respond to an unexpected acoustic stimulus with a jump. This ‘startle’ reflex can be reduced by a weak pre-stimulus (pre-pulse inhibition) such as a low-intensity tone or light. Schizophrenic patients exhibit less pre-pulse inhibition than control subjects. Drugs that mimic or release dopamine (e.g. apomorphine or amphetamine) as well as other drugs that induce schizophrenia-like behaviours (e.g. cannabinoids or phencyclidine) reduce pre-pulse inhibition in animals and antipsychotic drugs reverse this effect. Also, in a conditioned avoidance model, a rat may be trained to respond to a conditioned stimulus, such as a buzzer, by remaining immobile and thereby avoiding a painful shock; chlorpromazine impairs performance in this test, as well as in tests that demand active motor responses. In doses too small to reduce spontaneous motor activity, chlorpromazine reduces social interactions (grooming, mating, fighting, etc.) and also impairs performance in discriminant tests (e.g. requiring the animal to respond differently to red and green lights).
All first-generation antipsychotic drugs inhibit amphetamine-induced behavioural changes, reflecting their action on D2 receptors. Some atypical drugs have less activity on D2 receptors and are less active in such models, and also in the catalepsy model. They are, however, as efficacious as the older drugs in pre-pulse inhibition and conditioned avoidance tests. Both classic and atypical drugs, moreover, reduce the hyperactivity caused by phencyclidine (a glutamate antagonist; Ch. 37) in rodents. In humans, phencyclidine causes a schizophrenia-like syndrome. Conditioned avoidance and phencyclidine tests in animals may therefore be more appropriate guides to antipsychotic activity in humans.
In humans, antipsychotic drugs produce a state of apathy and reduced initiative. The recipient displays few emotions, is slow to respond to external stimuli and tends to drowse off. The subject is, however, easily aroused and can respond to questions accurately, with no marked loss of intellectual function. Aggressive tendencies are strongly inhibited. Effects differ from those of sedative anxiolytic drugs, which also cause drowsiness and confusion but with euphoria rather than apathy.
Many antipsychotic drugs are antiemetic (see Ch. 29), reflecting antagonism at dopamine, muscarinic, histamine and possibly 5-HT receptors.
Unwanted Effects
Extrapyramidal Motor Disturbances
Antipsychotic drugs produce two main kinds of motor disturbance in humans: acute dystonias and tardive dyskinesias, collectively termed extrapyramidal side effects. These all result directly or indirectly from D2 receptor blockade in the nigrostriatal pathway. Extrapyramidal side effects constitute one of the main disadvantages of first-generation antipsychotic drugs. The term atypical was originally applied to some of the newer compounds that show much less tendency to produce extrapyramidal side effects.
Acute dystonias are involuntary movements (restlessness, muscle spasms, protruding tongue, fixed upward gaze, torticollis [involuntary spasm of neck muscles]), often accompanied by symptoms of Parkinson’s disease (Ch. 39). They occur commonly in the first few weeks, often declining with time, and are reversible on stopping drug treatment. The timing is consistent with block of the dopaminergic nigrostriatal pathway. Concomitant block of muscarinic receptors and 5-HT2A receptors mitigates the motor effects of dopamine receptor antagonists (see above).
Tardive dyskinesia (see Klawans et al., 1988) develops after months or years (hence ‘tardive’) in 20–40% of patients treated with first-generation antipsychotic drugs, and is one of the main problems of antipsychotic therapy. Its seriousness lies in the fact that it is a disabling and often irreversible condition, which often gets worse when antipsychotic therapy is stopped and is resistant to treatment. The syndrome consists of involuntary movements, often of the face and tongue, but also of the trunk and limbs, which can be severely disabling. It resembles that seen after prolonged treatment of Parkinson’s disease with levodopa (see Ch. 39). The incidence depends greatly on drug, dose and age (being commonest in patients over 50).
There are several theories about the mechanism of tardive dyskinesia (see Casey, 1995). One is that it is associated with a gradual increase in the number of D2 receptors in the striatum, which is less marked during treatment with the atypical than with the first generation of antipsychotic drugs. Another possibility is that chronic block of inhibitory dopamine receptors enhances catecholamine and/or glutamate release in the striatum, leading to excitotoxic neurodegeneration (Ch. 39).
Drugs that rapidly dissociate from D2 receptors (e.g. clozapine, olanzapine, sertindole) induce less severe extrapyramidal side effects. A possible explanation for this (see Kapur & Seeman, 2001) is that with a rapidly dissociating compound, a brief surge of dopamine can effectively overcome the block by competition (see Ch. 2), whereas with a slowly dissociating compound, the level of block takes a long time to respond to the presence of endogenous dopamine, and is in practice non-competitive. Adverse motor effects may be avoided if fractional receptor occupation falls during physiological surges of dopamine. An extension of this idea is that perhaps a little D2 receptor activation may be beneficial. This could be produced, for example, by drugs that are D2 partial agonists (e.g. aripiprazole) in contrast to simple antagonists. It is thought that partial agonists reduce D2 hyperactivation in the mesolimbic pathway, thus alleviating positive symptoms of schizophrenia, but provide enough D2 receptor stimulation in the mesocortical pathway to prevent negative symptoms, and in the nigrostriatal pathway to prevent the development of extrapyramidal side effects. Newer D2 partial agonists such as bifeprunox are being developed, although questions about their efficacy and safety have arisen.
Antipsychotic-induced motor disturbances
Endocrine Effects
Dopamine, released in the median eminence by neurons of the tuberohypophyseal pathway (see Chs 32 and 38), acts physiologically via D2 receptors to inhibit prolactin secretion. Blocking D2 receptors by antipsychotic drugs can therefore increase the plasma prolactin concentration (Fig. 45.2), resulting in breast swelling, pain and lactation, which can occur in men as well as in women. As can be seen from Figure 45.2, the effect is maintained during chronic antipsychotic administration, without any habituation. Other less pronounced endocrine changes have also been reported, including a decrease of growth hormone secretion, but these, unlike the prolactin response, are believed to be relatively unimportant clinically.
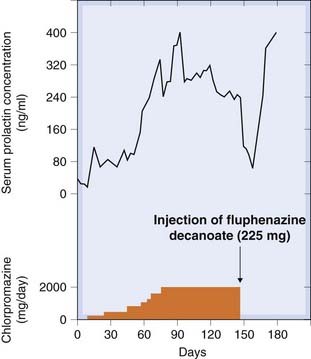
Fig. 45.2 Effects of antipsychotic drugs on prolactin secretion in a schizophrenic patient.
When daily dosage with chlorpromazine was replaced with a depot injection of fluphenazine, the plasma prolactin initally dropped, because of the delay in absorption, and then returned to a high level.
(From Meltzer H Y et al. 1978 In: Lipton et al. (eds) Psychopharmacology. A generation in progress. Raven Press, New York.)
Other Unwanted Effects
Drowsiness and sedation, which tend to decrease with continued use, occur with many antipsychotic drugs. Antihistamine (H1) activity is a property of some phenothiazine antipsychotics (e.g. chlorpromazine and methotrimeprazine) and contributes to their sedative and antiemetic properties (Ch. 38), but not to their antipsychotic action.
All antipsychotic drugs block a variety of receptors, particularly acetylcholine (muscarinic), histamine (H1), noradrenaline (α) and 5-HT (Table 45.1).
While block of muscarinic receptors produces a variety of peripheral effects, including blurring of vision and increased intraocular pressure, dry mouth and eyes, constipation and urinary retention (see Ch. 13), it may, however, also be beneficial in relation to extrapyramidal side effects (see above).
Blocking α-adrenoceptors causes orthostatic hypotension (see Ch. 14) but does not seem to be important for their antipsychotic action.
Weight gain is a common and troublesome side effect. Increased risk of diabetes and cardiovascular disease occurs with several atypical antipsychotic drugs. These effects are probably related to their antagonist actions at H1, 5-HT and muscarinic receptors.
Various idiosyncratic and hypersensitivity reactions can occur, the most important being the following:
Unwanted effects of antipsychotic drugs
Pharmacokinetic Aspects
Chlorpromazine, in common with other phenothiazines, is erratically absorbed after oral administration. Figure 45.3 shows the wide range of variation of the peak plasma concentration as a function of dosage in 14 patients. Among four patients treated at the high dosage level of 6–8 mg/kg, the variation in peak plasma concentration was nearly 90-fold; two showed marked side effects, one was well controlled and one showed no clinical response.
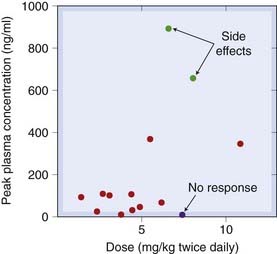
Fig. 45.3 Individual variation in the relation between dose and plasma concentration of chlorpromazine in a group of schizophrenic patients.
(Data from Curry S H et al. 1970 Arch Gen Psychiatry 22: 289.)
The relationship between the plasma concentration and the clinical effect of antipsychotic drugs is highly variable, and the dosage has to be adjusted on a trial-and-error basis. This is made even more difficult by the fact that at least 40% of schizophrenic patients fail to take drugs as prescribed. It is remarkably fortunate that the acute toxicity of antipsychotic drugs is slight, given the unpredictability of the clinical response.
The plasma half-life of most antipsychotic drugs is 15–30 h, clearance depending entirely on hepatic transformation by a combination of oxidative and conjugative reactions.
Most antipsychotic drugs can be given orally or in urgent situations by intramuscular injection. Slow-release (depot) preparations of many are available, in which the active drug is esterified with heptanoic or decanoic acid and dissolved in oil. Given as an intramuscular injection, the drug acts for 2–4 weeks, but initially may produce acute side effects. These preparations are widely used to minimise compliance problems.
Clinical Use and Clinical Efficacy
The major use of antipsychotic drugs is in the treatment of schizophrenia and acute behavioural emergencies, but they are also used to treat other conditions, such as deviant antisocial behaviour, motor tics and intractable hiccup. Their use to treat restlessness and agitation in the elderly is highly questionable. In addition, they are used as adjunct therapy in psychotic depression, bipolar disorder and mania. Some of the newer antipsychotic drugs (e.g. sulpiride) have been claimed to have specific antidepressant actions. Phenothiazines and related drugs are also useful as antiemetics (see Ch. 29). Minor uses include the treatment of Huntington’s chorea (mainly haloperidol; see Ch. 39).
The clinical efficacy of antipsychotic drugs in enabling schizophrenic patients to lead more normal lives has been demonstrated in many controlled trials. The inpatient population (mainly chronic schizophrenics) of mental hospitals declined sharply in the 1950s and 1960s. The efficacy of the newly introduced antipsychotic drugs was a significant enabling factor, as well as the changing public and professional attitudes towards hospitalisation of the mentally ill.
Antipsychotic drugs, apart from their side effects, have two main shortcomings:
The newer atypical antipsychotic drugs may overcome these shortcomings to some degree. However, a recent meta-analysis (Leucht et al., 2009) concluded that, of the atypical antipsychotic drugs examined, only amisulpride, clozapine, olanzapine and risperidone were better than first-generation antipsychotic drugs for overall efficacy. The other atypical drugs were not more efficacious than the first-generation drugs, even for negative symptoms.
Clinical uses of antipsychotic drugs 
Future Developments
Preclinical and clinical studies have provided encouraging evidence that agonists of group II metabotropic glutamate receptors (mGluR2 and mGluR3; see Ch. 37) are effective in the treatment of the positive symptoms of schizophrenia (Patil et al., 2007). Furthermore, agonists at mGluR5 receptors may improve positive and negative symptoms as well as cognitive function. It may also be possible to enhance the action of endogenous glutamate at these receptors with positive allosteric modulator drugs. For more detailed information, see Conn et al. (2009).
References and Further Reading
Stahl S.M. Antipsychotics and mood stabilizers, third ed. New York: Cambridge University Press; 2008. (Highly readable, yet detailed, description of the biology of schizophrenia and of the mechanisms of action of the drugs used to treat the disorder)
Craddock N., O’Donovan M.C., Owen M.J. The genetics of schizophrenia and bipolar disorder: dissecting psychosis. J. Med. Genet.. 2005;42:193-204.
Harrison P.J. Schizophrenia: a disorder of development. Curr. Opin. Neurobiol.. 1997;7:285-289. (Reviews persuasively the evidence favouring abnormal early brain development as the basis of schizophrenia)
Harrison P.J., Weinberger D.R. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry. 2005;10:40-68.
Sim K., Cullen T., Ongur D., Heckers S. Testing models of thalamic dysfunction in schizophrenia using neuroimaging. J. Neural. Transm.. 2006;113:907-928.
van Haren N.E., Hulshoff Pol H.E., Schnack H.G., et al. Focal gray matter changes in schizophrenia across the course of the illness: a 5-year follow-up study. Neuropsychopharmacology. 2007;32:2057-2066.
Dopamine, glutamate and 5-hydroxytryptamine
Bridges T.M., Williams R., Lindsley C.W. Design of potent GlyT1 inhibitors: in vitro and in vivo profiles. Curr. Opin. Mol. Ther.. 2008;10:591-601. (Discusses the potential of reducing glycine uptake as a means of enhancing activation of NMDA receptors)
Busatto G.F., Kerwin R.W. Perspectives on the role of serotonergic mechanisms in the pharmacology of schizophrenia. J. Psychopharmacol.. 1997;11:3-12. (Assesses the evidence implicating 5-HT as well as dopamine in the action of antipsychotic drugs)
Conn P.J., Lindsley C.W., Jones C.K. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol. Sci.. 2009;30:25-31. (Describes an exciting new area in the development of novel antipsychotic drugs)
Coyle J.T. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell. Mol. Neurobiol.. 2006;26:365-384. (Describes the emerging view of the importance of glutamate in schizophrenia)
Javitt D.C. Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry. 2004;9:984-997. (Summarises the evidence favouring disturbed glutamate function in schizophrenia, including data from recent clinical trials)
Laruelle M., Abi-Dargham A., Gil R., et al. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol. Psychiatry. 1999;46:56-72. (The first direct evidence for increased dopamine function as a cause of symptoms in schizophrenia)
Moghaddam B. Bringing order to the glutamate chaos in schizophrenia. Neuron. 2003;40:861-864. (Reviews evidence from recent genetic findings, suggesting the abnormalities in glutamate transmission may play a key role in schizophrenia)
Patil S.T., Zhang L., Martenyi F., et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat. Med.. 2007;13:1102-1107.
Seeman P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse. 1987;1:133-152. (Convincing and widely quoted review of role of dopamine receptors in schizophrenia)
Shim S.S., Hammonds M.D., Kee B.S. Potentiation of the NMDA receptor in the treatment of schizophrenia: focused on the glycine site. Eur. Arch. Psych. Clin. Neurosci.. 2008;258:16-27. (Review of recent clinical trials of agents that enhance the action of glycine at the NMDA receptor)
Toda M., Abi-Dargham A. Dopamine hypothesis of schizophrenia: making sense of it all. Curr. Psychiatry Rep.. 2007;9:329-336.
Basile V.S., Masellis M., Potkin S.G., Kennedy J.L. Pharmacogenomics in schizophrenia: the quest for individualized therapy. Hum. Mol. Genet.. 2002;11:2517-2530. (Review of inconclusive evidence for association between clozapine responsiveness and gene polymorphisms)
Kapur S., Seeman P. Does fast dissociation from the dopamine D2 receptor explain the action of atypical antipsychotics? A new hypothesis. Am. J. Psychiatry. 2001;158:360-369. (Suggests that differences in dissociation rates, rather than receptor selectivity profiles, may account for differing tendency of drugs to cause motor side effects)
Kapur S., Arenovich T., Agid O., et al. Evidence for onset of antipsychotic effects within the first 24 hours of treatment. Am. J. Psychiatry. 2005;162:939-946.
Leucht S., Busch R., Hamann J., Kissling W., Kane J.M. Early-onset hypothesis of antipsychotic drug action: a hypothesis tested, confirmed and extended. Biol. Psychiatry. 2005;57:1543-1549.
Leucht S., Corves C., Arbter D., et al. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373:31-41. (A comparison of the clinical effectivness of new and old antipsychotic drugs)
Ögren S.O. The behavioural pharmacology of typical and atypical antipsychotic drugs. In: Csernasky J.G., editor. Antipsychotics. Handbook of experimental pharmacology, vol. 120. Berlin: Springer; 1996.
Remington G. Understanding antipsychotic ‘atypicality’: a clinical and pharmacological moving target. J. Psychiatry Neurosci.. 2003;28:275-284. (An informative critique of the basis on which antipsychotic drugs are classified)
Casey D.E. Tardive dyskinesia: pathophysiology. In: Bloom F.E., Kupfer D.J., editors. Psychopharmacology: a fourth generation of progress. New York: Raven Press, 1995.
Klawans H.L., Tanner C.M., Goetz C.G. Epidemiology and pathophysiology of tardive dyskinesias. Adv. Neurol.. 1988;49:185-197.
Pisani A., Bernardi G., Ding J., Surmeier D.J. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci.. 2007;30:545-553.
1In this respect, the study of schizophrenia lags some years behind that of Alzheimer’s disease (Ch. 39), where understanding of the pathogenesis has progressed rapidly to the point where promising new drug targets can be identified. On the other hand, pragmatists can argue that drugs against Alzheimer’s disease are so far only marginally effective, whereas current antipsychotic drugs deliver great benefits, even though we do not quite know how they work.
2Schizophrenia is a condition where the patient exhibits symptoms of psychosis (e.g. delusions, hallucinations and disorganised behaviour). Psychotic episodes may also occur as a result of taking certain recreational drugs (see Ch. 47); as an adverse effect of drug treatment, for example steroid-induced psychoses; or in disorders such as mania, depression (see Ch. 46) and Alzheimer’s disease (see Ch. 39).
3Kraepelin, who first described the condition, used the term dementia praecox (premature dementia) to describe the cognitive impairment associated with schizophrenia.
4There are, however, exceptions to this simple rule. Up to one-third of schizophrenic patients fail to respond even when D2 receptor blockade exceeds 90%, and clozapine (see Table 45.1) can be effective at much lower levels of block.
5An increase in dopamine receptor density in schizophrenia has been reported in some studies, but not consistently, and the interpretation is complicated by the fact that chronic antipsychotic drug treatment is known to increase dopamine receptor expression.
6The D4 receptor attracted attention on account of the high degree of genetic polymorphism that it shows in human subjects, and because some of the newer antipsychotic drugs (e.g. clozapine) have a high affinity for this receptor subtype. However, a specific D4-receptor antagonist proved ineffective in clinical trials.
7Early antipsychotic drugs (e.g. chlorpromazine) had actions at various receptors but also had unwanted side effects that resulted from activity at other receptors. Towards the end of the 20th century, drug development, not just of antipsychotic drugs, was focused largely on developing agents with a single action with the intention of reducing unwanted side effects. This philosophy drove the search for selective D4 receptor antagonists, which proved ineffective. What is now apparent is that drugs with selected multiple actions (e.g. a combination of D2 antagonism and 5-HT2A antagonism) may have a better therapeutic profile.