46 Antidepressant drugs
Overview
Depression is an extremely common psychiatric condition, about which a variety of neurochemical theories exist, and for which a corresponding variety of different types of drug are used in treatment. It is a field in which therapeutic empiricism has led the way, with mechanistic understanding tending to lag behind, part of the difficulty being that animal models cannot address the mood changes that define the human condition. In this chapter, we discuss the current understanding of the nature of the disorder, and describe the major drugs that are used to treat it.
The Nature of Depression
Depression is the most common of the affective disorders (defined as disorders of mood rather than disturbances of thought or cognition); it may range from a very mild condition, bordering on normality, to severe (psychotic) depression accompanied by hallucinations and delusions. Worldwide, depression is a major cause of disability and premature death. In addition to the significant suicide risk, depressed individuals are more likely to die from other causes, such as heart disease or cancer. Depression is a heterogeneous disorder with patients presenting with one or more core symptoms and depression is often associated with other psychiatric conditions including anxiety, eating disorders and drug addiction.
The symptoms of depression include emotional and biological components. Emotional symptoms include:
There are two distinct types of depressive syndrome, namely unipolar depression, in which the mood changes are always in the same direction, and bipolar affective disorder, in which depression alternates with mania. Mania is in most respects exactly the opposite, with excessive exuberance, enthusiasm and self-confidence, accompanied by impulsive actions, these signs often being combined with irritability, impatience and aggression, and sometimes with grandiose delusions of the Napoleonic kind. As with depression, the mood and actions are inappropriate to the circumstances.
Unipolar depression is commonly (about 75% of cases) non-familial, clearly associated with stressful life events, and usually accompanied by symptoms of anxiety and agitation; this type is sometimes termed reactive depression. Other cases (about 25%, sometimes termed endogenous depression) show a familial pattern, unrelated to obvious external stresses, and with a somewhat different symptomatology. This distinction is made clinically, but there is little evidence that antidepressant drugs show significant selectivity between these conditions.
Bipolar depression, which usually appears in early adult life, is less common and results in oscillating depression and mania over a period of a few weeks. It can be difficult to differentiate between mild bipolar depression and unipolar depression. Also, bipolar manic episodes can be confused with episodes of psychosis (see Ch. 45). There is a strong hereditary tendency, but no specific susceptibility genes have been identified either by genetic linkage studies of affected families, or by comparison of affected and non-affected individuals.
Depression cannot be attributed to altered neuronal activity within a single brain region. Brain imaging studies have indicated that the prefrontal cortex, amygdala and hippocampus may all be involved in different components of these disorders.
Theories of Depression
The Monoamine Theory
The main biochemical theory of depression is the monoamine hypothesis, first proposed by Schildkraut in 1965, which states that depression is caused by a functional deficit of the monoamine transmitters, noradrenaline and 5-hydroxytryptamine (5-HT) at certain sites in the brain, while mania results from a functional excess. For reviews of the evolving status of the theory, see Maes & Meltzer (1995) and Manji et al. (2001).
The monoamine hypothesis grew originally out of associations between the clinical effects of various drugs that cause or alleviate symptoms of depression and their known neurochemical effects on monoaminergic transmission in the brain. This pharmacological evidence, which is summarised in Table 46.1, gives general support to the monoamine hypothesis, although there are several anomalies. Attempts to obtain more direct evidence, by studying monoamine metabolism in depressed patients or by measuring changes in the number of monoamine receptors in postmortem brain tissue, have tended to give inconsistent and equivocal results, and the interpretation of these studies is often problematic, because the changes described are not specific to depression. Similarly, investigation by functional tests of the activity of known monoaminergic pathways (e.g. those controlling pituitary hormone release) in depressed patients have also given equivocal results.
Table 46.1 Pharmacological evidence supporting the monoamine hypothesis of depression
| Drug(s) | Principal action | Effect in depressed patients |
|---|---|---|
| Tricyclic antidepressants | Block noradrenaline and 5-HT reuptake | Mood ↑ |
| Monoamine oxidase (MAO) inhibitors | Increase stores of noradrenaline and 5-HT | Mood ↑ |
| Reserpine | Inhibits noradrenaline and 5-HT storage | Mood ↓ |
| α-Methyltyrosine | Inhibits noradrenaline synthesis | Mood ↓ (calming of manic patients) |
| Methyldopa | Inhibits noradrenaline synthesis | Mood ↓ |
| Electroconvulsive therapy | ? Increases central nervous system responses to noradrenaline and 5-HT | Mood ↑ |
| Tryptophan (5-hydroxytryptophan) | Increases 5-HT synthesis | Mood ? ↑ in some studies |
| Tryptophan depletion | Decreases brain 5-HT synthesis | Induces relapse in SSRI-treated patients |
5-HT, 5-hydroxytryptamine; SSRI, selective serotonin reuptake inhibitor.
The pharmacological evidence does not enable a clear distinction to be drawn between the noradrenaline and 5-HT theories of depression. Clinically, it seems that inhibitors of noradrenaline reuptake and of 5-HT reuptake are equally effective as antidepressants (see below), although individual patients may respond better to one or the other.
Other evidence in support of the monoamine theory is that agents known to block noradrenaline or 5-HT synthesis consistently reverse the therapeutic effects of antidepressant drugs that act selectively on these two transmitter systems (see Table 46.1).
Any theory of depression has to take account of the fact that the direct neurochemical effects of antidepressant drugs appear very rapidly (minutes to hours), whereas their antidepressant effects take weeks to develop. A similar situation exists in relation to antipsychotic drugs (Ch. 45) and some anxiolytic drugs (Ch. 43), suggesting that the secondary, adaptive changes in the brain, rather than the primary drug effect, are responsible for the clinical improvement. Rather than thinking of the monoamine deficiency as causing direct changes in the activity of putative ‘happy’ or ‘sad’ neurons in the brain, we should think of the monoamines as regulators of longer-term trophic effects, whose time course is paralleled by mood changes.
With improved neuroimaging methods for studying neurotransmitter function in the living human brain, as described in Chapter 36, our understanding of the causes of depression and how drugs can alleviate depression should improve.
Neuroendocrine Mechanisms
Various attempts have been made to test for a functional deficit of monoamine pathways in depression. Hypothalamic neurons controlling pituitary function receive noradrenergic and 5-HT inputs, which control the discharge of these cells. Hypothalamic cells release corticotrophin-releasing hormone (CRH), which stimulates pituitary cells to secrete adrenocorticotrophic hormone (ACTH), leading in turn to cortisol secretion. The plasma cortisol concentration is usually high in depressed patients, and it fails to respond with the normal fall when a synthetic steroid, such as dexamethasone, is given. This formed the basis of a clinical test, the dexamethasone suppression test (also used in the diagnosis of Cushing’s syndrome; see Ch. 32). Other hormones in plasma are also affected, for example growth hormone concentration is reduced and prolactin is increased. While these changes are consistent with deficiencies in monoamine transmission, they are not specific to depressive syndromes.
Corticotrophin-releasing hormone is widely distributed in the brain and has behavioural effects that are distinct from its endocrine functions. Injected into the brain of experimental animals, CRH mimics some effects of depression in humans, such as diminished activity, loss of appetite and increased signs of anxiety. Furthermore, CRH concentrations in the brain and cerebrospinal fluid of depressed patients are increased. Therefore CRH hyperfunction, as well as monoamine hypofunction, may be associated with depression (see Holsboer, 1999). Raised CRH levels are associated with stress and, in many cases, depression is preceded by periods of chronic stress.
Trophic Effects and Neuroplasticity
It has been suggested that lowered levels of brain-derived neurotrophic factor (BDNF) or malfunction of its receptor, TrkB, plays a significant role in the pathology of this condition. Depressive behaviour is often associated with a reduction in BDNF expression and treatment with antidepressants elevates BDNF levels.
Changes in glutamatergic neurotransmission may also be involved in depression. Sufferers from depression have been shown to have elevated cortical levels of glutamate. Antidepressant treatment may reduce glutamate release and depress NMDA receptor function. The effects of antidepressants on activity-induced long-term potentiation (LTP; see Ch. 37) at hippocampal glutamatergic synapses is complex—both depression and facilitation have been observed and may occur with acute antidepressant administration, thus calling into question the relevance to the therapeutic response.
Another view (see Charney & Manji, 2004; Duman, 2004; Racagni & Popoli, 2008) is that major depression is associated with neuronal loss in the hippocampus and prefrontal cortex, and that antidepressant therapies of different kinds act by inhibiting or actually reversing this loss by stimulating neurogenesis.1 This surprising idea is supported by various lines of evidence:
Figure 46.1 summarises the possible mechanisms involved. It should be stressed that these hypotheses are far from proven, but the diagram emphasises the way in which the field has moved on since the formulation of the monoamine hypothesis, suggesting a range of possible targets for the next generation of antidepressant drugs.2
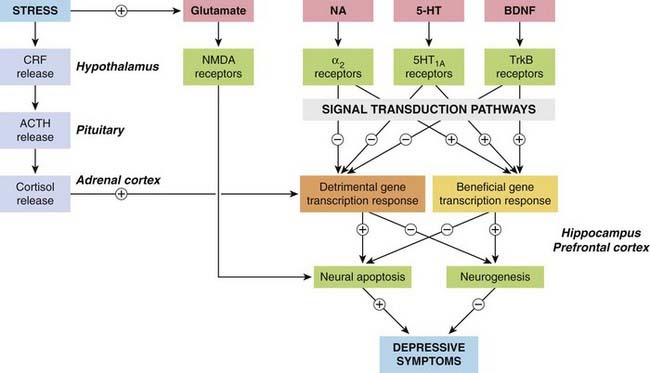
Fig. 46.1 Simplified diagram showing mechanisms believed to be involved in the pathophysiology of depression.
The main prodepressive pathways involve the hypothalamic–pituitary–adrenal axis, which is activated by stress and in turn enhances the excitotoxic action of glutamate, mediated by NMDA receptors (see Ch. 37), and switches on the expression of genes that promote neural apoptosis in the hippocampus and prefrontal cortex. The antidepressive pathways involve the monoamines noradrenaline (NA) and 5-hydroxytryptamine (5-HT), which act on G-protein-coupled receptors, and the brain-derived neurotrophic factor (BDNF), which acts on a kinase-linked receptor (TrkB), switching on genes that protect neurons against apoptosis and also promote neurogenesis. For further detail, see Charney & Manji (2004). ACTH, adrenocorticotrophic hormone; CRF, corticotrophin-releasing factor.
Monoamine theory of depression 
Antidepressant Drugs
Types of Antidepressant Drug
Antidepressant drugs fall into the following categories.
Inhibitors of monoamine uptake
Monoamine oxidase inhibitors (MAOIs)
Table 46.2 summarises the main features of these types of drug. Recent updates (Bosker et al., 2004; Pacher & Kecsemeti, 2004; Stahl, 2008) provide more detail. Mention should also be made of electroconvulsive therapy (ECT), electromagnetic therapy, deep brain stimulation and vagus stimulation, which are effective and usually act more rapidly than antidepressant drugs (see later section).
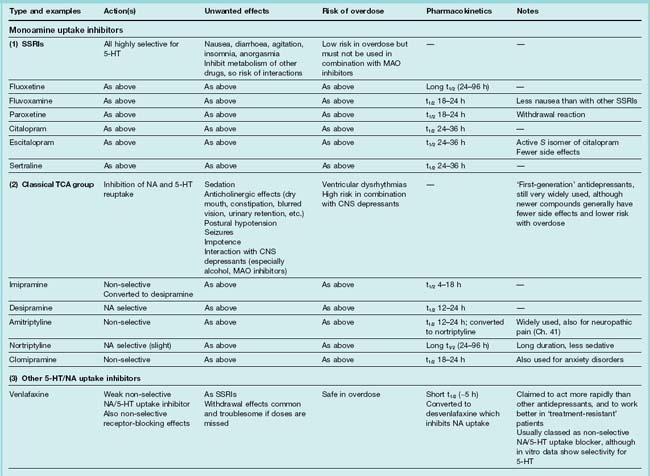
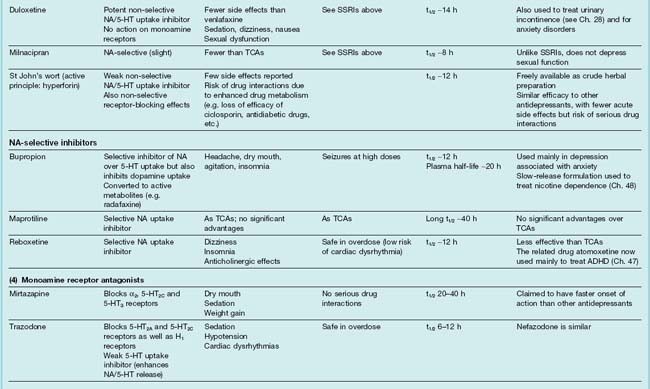
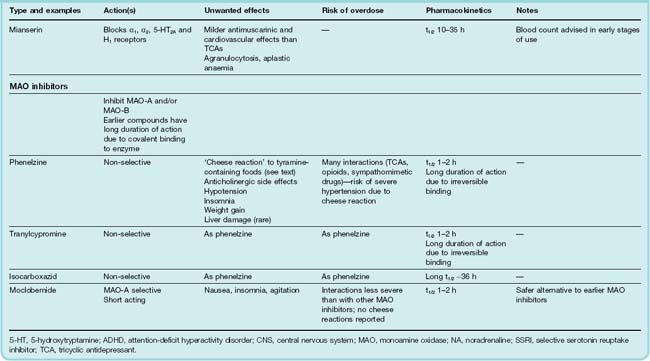
Types of antidepressant drugs
Testing of Antidepressant Drugs
Animal Models
Progress in unravelling the neurochemical mechanisms is, as in so many areas of psychopharmacology, limited by the lack of good animal models of the clinical condition. There is no known animal condition corresponding to the inherited condition of depression in humans, but various procedures have been described that produce in animals behavioural states (withdrawal from social interaction, loss of appetite, reduced motor activity, etc.) typical of human depression (see Table 46.3 and review by Cryan & Slattery, 2007). The use of genetically modified mice to mimic various aspects of the disorder may provide interesting models (see Gardier, 2009). However, the similarity of these animal models to human depression is questionable.
Table 46.3 Animal models used to study depression
| Model | Description |
|---|---|
| Forced swim test (Porsolt test) | Classical model for antidepressant efficacy. Rodents are placed in an inescapable container of water on two occasions. On the second test, acute antidepressant drugs increase the escape behaviour Provides good assessment of efficacy for monoaminergic antidepressant drugs. Effects are seen after acute treatment unlike the delayed effects seen in humans |
| Modified swim test | Same basic test as above but separates swimming versus climbing behaviour to dissociate between serotonergic and catecholaminergic activity |
| Tail suspension test | Primarily used for mice. The animal is suspended from the tail and the time to an immobile posture recorded |
| Learned helplessness | Rodents are exposed to repeated inescapable foot shock resulting in a failure to subsequently escape when able to Antidepressant drugs increase the number of escapes after conditioning. Acute effects with antidepressants are observed but not all animals develop the response |
| Olfactory bulbectomy | Removal of the olfactory bulbs in rats causes behavioural and neurochemical changes that reflect symptoms observed in depressed subjects. Responds to chronic antidepressant treatment |
| Maternal deprivation | Pups are removed from the dam for brief periods early postnatal which changes the maternal care of the offspring. The offspring go on to develop a phenotype that expresses behavioural, neurochemical and biochemical changes that reflect aspects of depression. Not all animals develop these changes |
| Chronic mild stress | Animals are subjected to a sequence of stressors over a period of ∼14 days. The stressors differ each day forming a period of unpredictable chronic stress. The animals develop a range of behavioural, neurochemical and biochemical changes that reflect symptoms seen in depression. Responds to chronic antidepressant treatment |
Tests on Humans
Clinically, the effect of antidepressant drugs is usually measured by a subjective rating scale such as the 17-item Hamilton Rating Scale. Clinical depression takes many forms, and the symptoms vary between patients and over time. Quantitation is therefore difficult, and the many clinical trials of antidepressants have generally shown rather weak effects, after allowance for quite large placebo responses. There is also a high degree of individual variation, with 30–40% of patients failing to show any improvement, possibly due to genetic factors (see later section on Clinical Effectiveness).
Mechanism of Action of Antidepressant Drugs
Chronic Adaptive Changes
Given the discrepancy between the fast onset of the neurochemical effects of antidepressant drugs and the slow onset of their antidepressant effects, efforts have been made to determine whether the therapeutic benefits arise from slow adaptive changes induced by chronic exposure to these drugs (Racagni & Popoli, 2008).
This approach led to the discovery that certain monoamine receptors, in particular β1- and α2 adrenoceptors, are consistently downregulated following chronic antidepressant treatment and, in some cases, by electroconvulsive therapy too. This can be demonstrated in experimental animals as a reduction in the number of binding sites, as well as by a reduction in the functional response to agonists (e.g. stimulation of cAMP formation by β-adrenoceptor agonists). Receptor downregulation probably also occurs in humans, because endocrine responses to clonidine, an α2 adrenoceptor agonist, are reduced by long-term antidepressant treatment. However, the relevance of these findings to the antidepressant response is unclear. Loss of β-adrenoceptors as a factor in alleviating depression does not fit comfortably with theory, because β-adrenoceptor antagonists are not antidepressant.
On acute administration, one would expect inhibition of 5-HT uptake (e.g. by SSRIs) to increase the level of 5-HT in the synapse by inhibiting reuptake into the nerve terminals. However, the increase in synaptic 5-HT levels has been observed to be less than expected. This is because increased activation of 5-HT1A receptors on the soma and dendrites of 5-HT-containing raphe neurons (Fig. 46.2A) inhibits these neurons and thus reduces 5-HT release, thus cancelling out to some extent the effect of inhibiting reuptake into the terminals. On prolonged drug treatment, the elevated level of 5-HT in the somatodendritic region desensitises the 5-HT1A receptors, reducing their inhibitory effect on 5-HT release from the nerve terminals.
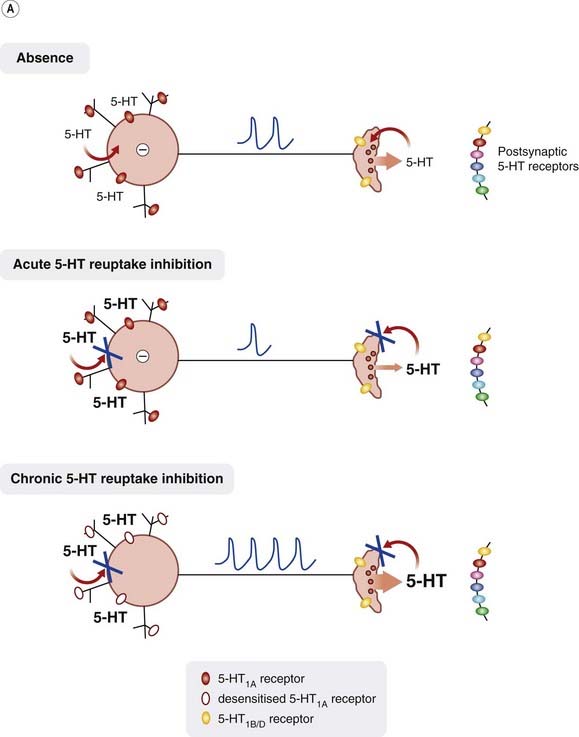
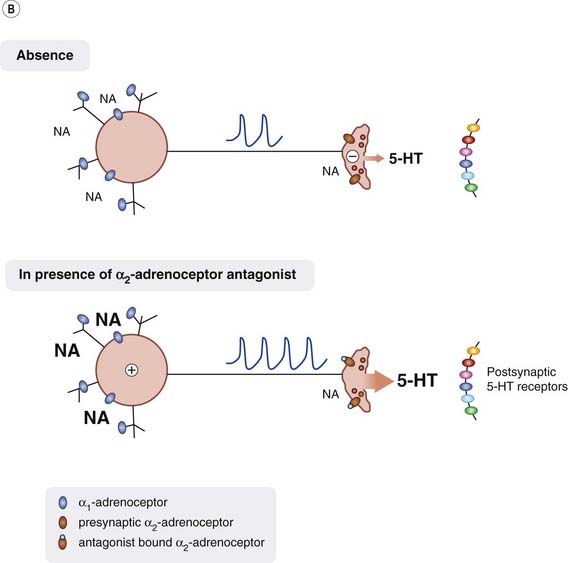
Fig. 46.2 Control of 5-HT release.
[A] 5-HT release is controlled by the inhibitory action of 5-HT on somatodendritic 5-HT1A receptors. Acute inhibition of 5-HT reuptake results in increased extracellular levels of 5-HT but this increases somatodendritic 5-HT1A receptor-mediated inhibition, hence synaptic 5-HT levels do not rise as much as expected. 5-HT1A receptors eventually desensitise, resulting in reduced inhibition and thus greater 5-HT release. [B] 5-HT release is controlled by both an excitatory action of noradrenaline (NA) on somatodendritic α1-adrenoceptors and an inhibitory action on α2 adrenoceptors on serotonergic nerve terminals. Block of α2 adrenoceptors located on noradrenergic neurons (not shown) enhances noradrenaline release resulting in further excitation of serotonergic neurons, while block of α2 adrenoceptors on serotonergic neurons removes presynaptic inhibition and thus 5-HT release is enhanced.
The need to desensitise somatodendritic 5-HT1A receptors could thus explain the slow onset of antidepressant action of 5-HT uptake inhibitors. Rather than reduce receptor function by desensitisation, it should be possible to produce the same effect simply by blocking the receptors with an antagonist. Pindolol, a non-selective β-adrenoceptor blocker, which also has affinity for 5-HT1A receptors, has been used in conjunction with 5-HT uptake inhibitors to speed up the onset of antidepressant action (see Ballasteros & Callado, 2004). However, drugs with combined 5-HT1A antagonism and SSRI properties have been developed but have not been found to be effective in man, perhaps because they block both 5-HT1A autoreceptors and postsynaptic 5-HT1A receptors, the latter effect occluding the beneficial effect of the former.
Noradrenergic Control of 5-HT Release
Block of presynaptic α2 autoreceptors on noradrenergic nerve terminals throughout the CNS will reduce the negative feedback from released noradrenaline and thus enhance further noradrenaline release (see Chs 14 and 36). In addition, α2 adrenoceptor antagonists can indirectly enhance 5-HT release. This can occur in several ways (see Fig. 46.2B):
The effect of α2 adrenoceptor antagonists on synaptic noradrenaline and 5-HT levels would be rapid in onset and so these changes must somehow induce other, slower adaptive responses that give rise to the slowly developing antidepressant effects.
Gene Expression and Neurogenesis
More recently, interest has centred on intracellular signalling pathways, changes in gene expression and neurogenesis. Much attention has been focused on how antidepressants may activate the transcription factor, CREB, a cAMP response element binding protein (see Ch. 48). The role of other transcription factors such as those of the Fos family and NF-κB have been less extensively studied. As described above, several antidepressant drugs appear to promote neurogenesis in the hippocampus, a mechanism that could account for the slow development of the therapeutic effect. The role of raised synaptic noradrenaline and 5-HT levels in inducing changes in gene expression and neurogenesis, and the mechanisms involved, await further elucidation.
Monoamine Uptake Inhibitors
Selective 5-Hydroxytryptamine Uptake Inhibitors
Drugs of this type (often termed selective serotonin reuptake inhibitors or SSRIs) include fluoxetine, fluvoxamine, paroxetine, citalopram, escitalopram and sertraline (see Table 46.2). They are the most commonly prescribed group of antidepressants. As well as showing selectivity with respect to 5-HT over noradrenaline uptake (Fig. 46.3), they are less likely than TCAs to cause anticholinergic side effects and are less dangerous in overdose. In contrast to MAOIs, they do not cause ‘cheese reactions’. They are as effective as TCAs and MAOIs in treating depression of moderate degree, but probably less effective than TCAs in treating severe depression. They are also used to treat anxiety disorders (see Ch. 43).
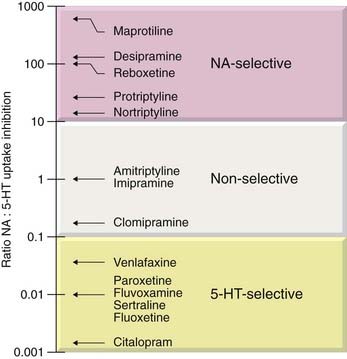
Fig. 46.3 Selectivity of inhibition of noradrenaline and 5-hydroxytryptamine uptake by various antidepressants.
Individual patients may respond more favourably to one SSRI than another. This may reflect other, pharmacological properties of each individual drug as none is devoid of other actions. Fluoxetine has 5-HT2C antagonist activity, a property it shares with other non-SSRI antidepressants such as mirtazapine. This may also contribute to its therapeutic effect in the treatment of anorexia and bulimia. Sertraline is a weak inhibitor of dopamine uptake. Escitalopram is the S isomer of racemic citalopram. It lacks the antihistamine and CYP2D6 inhibitory properties of the R isomer.
Pharmacokinetic aspects
The SSRIs are well absorbed, and most have plasma half-lives of 18–24 h (fluoxetine is longer acting: 24–96 h). The delay of 2–4 weeks before the therapeutic effect develops is similar to that seen with other antidepressants. Paroxetine and fluoxetine are not used in combination with TCAs, whose hepatic metabolism they inhibit through an interaction with CYP2D6, for fear of increasing TCA toxicity.
Unwanted effects
Common side effects include nausea, anorexia, insomnia, loss of libido and failure of orgasm.4 Some of these unwanted effects result from the enhanced stimulation of postsynaptic 5-HT receptors as a result of the drugs increasing the levels of extracellular 5-HT. This can be either stimulation of the wrong type of 5-HT receptor (e.g. 5-HT2, 5-HT3 and 5-HT4 receptors) or stimulation of the same receptor that gives therapeutic benefit (e.g. postsynaptic 5-HT1A receptors) but in the wrong brain region (i.e. enhanced stimulation of 5-HT receptors can result in both therapeutic and adverse responses).
In combination with MAOIs, SSRIs can cause a ‘serotonin syndrome’ characterised by tremor, hyperthermia and cardiovascular collapse, from which deaths have occurred.
There have been reports of increased aggression, and occasionally violence, in patients treated with fluoxetine, but these have not been confirmed by controlled studies. The use of SSRIs is not recommended for treating depression in children under 18, in whom efficacy is doubtful and adverse effects, including excitement, insomnia and aggression in the first few weeks of treatment, may occur. The possibility of increased suicidal ideation is a concern in this age group (see below).
Despite the apparent advantages of 5-HT uptake inhibitors over TCAs in terms of side effects, the combined results of many trials show little overall difference in terms of patient acceptability (Song et al., 1993; Cipriani et al., 2009).
5-HT uptake inhibitors are used in a variety of other psychiatric disorders, as well as in depression, including anxiety disorders and obsessive compulsive disorder (see Ch. 43).
Selective serotonin reuptake inhibitors (SSRIs)
Tricyclic Antidepressant Drugs
Tricyclic antidepressants (TCAs; imipramine, desipramine, amitriptyline, nortriptyline, clomipramine) are still widely used. They are, however, far from ideal in practice, and it was the need for drugs that act more quickly and reliably, produce fewer side effects and are less hazardous in overdose that led to the introduction of newer 5-HT reuptake inhibitors and other antidepressants.
TCAs are closely related in structure to the phenothiazines (Ch. 45) and were initially synthesised (in 1949) as potential antipsychotic drugs. Several are tertiary amines, with two methyl groups attached to the basic nitrogen atom. They are quite rapidly demethylated in vivo (Fig. 46.4) to the corresponding secondary amines (e.g. imipramine to desipramine, amitriptyline to nortriptyline), which are themselves active and may be administered as drugs in their own right. Other tricyclic derivatives with slightly modified bridge structures include doxepin. The pharmacological differences between these drugs are not very great and relate mainly to their side effects, which are discussed below.
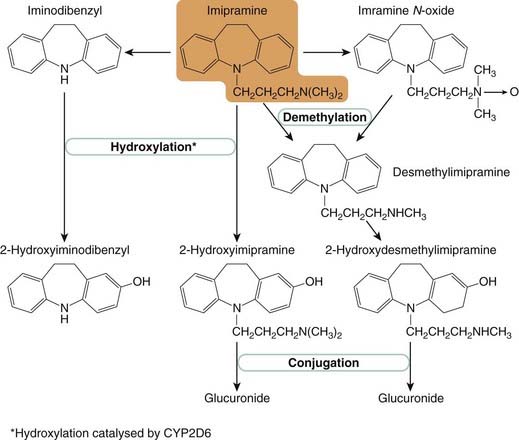
Fig. 46.4 Metabolism of imipramine, which is typical of that of other tricyclic antidepressants.
The hydroxylating enzyme, CYP2D6, is subject to genetic polymorphism, which may account for individual variation in response to tricyclic antidepressants (see Ch. 11).
TCAs are also used to treat neuropathic pain (see Ch. 41).
Mechanism of action
As discussed above, the main immediate effect of TCAs is to block the uptake of amines by nerve terminals, by competition for the binding site of the amine transporter (Ch. 14). Most TCAs inhibit noradrenaline and 5-HT uptake (Fig. 46.3) but have much less effect on dopamine uptake. It has been suggested that improvement of emotional symptoms reflects mainly an enhancement of 5-HT-mediated transmission, whereas relief of biological symptoms results from facilitation of noradrenergic transmission. Interpretation is made difficult by the fact that the major metabolites of TCAs have considerable pharmacological activity (in some cases greater than that of the parent drug) and often differ from the parent drug in respect of their noradrenaline/5-HT selectivity (Table 46.4).
Table 46.4 Inhibition of neuronal noradrenaline and 5-HT uptake by tricyclic antidepressants and their metabolites
| Drug/metabolite | NA uptake | 5-HT uptake |
|---|---|---|
| Imipramine | +++ | ++ |
| Desmethylimipramine (DMI) | ++++ | + |
| Hydroxy-DMI | +++ | − |
| Clomipramine (CMI) | ++ | +++ |
| Desmethyl-CMI | +++ | + |
| Amitriptyline (AMI) | ++ | ++ |
| Nortriptyline (desmethyl-AMI) | +++ | ++ |
| Hydroxynortriptyline | ++ | ++ |
In addition to their effects on amine uptake, most TCAs affect other receptors, including muscarinic acetylcholine receptors, histamine receptors and 5-HT receptors. The antimuscarinic effects of TCAs do not contribute to their antidepressant effects but are responsible for various side effects.
Unwanted effects
In non-depressed human subjects, TCAs cause sedation, confusion and motor incoordination. These effects occur also in depressed patients in the first few days of treatment, but tend to wear off in 1–2 weeks as the antidepressant effect develops.
Tricyclic antidepressants produce a number of troublesome side effects, mainly due to interference with autonomic control.
Atropine-like effects include dry mouth, blurred vision, constipation and urinary retention. These effects are strong with amitriptyline and much weaker with desipramine. Postural hypotension occurs with TCAs. This may seem anomalous for drugs that enhance noradrenergic transmission, and possibly results from an effect on adrenergic transmission in the medullary vasomotor centre. The other common side effect is sedation, and the long duration of action means that daytime performance is often affected by drowsiness and difficulty in concentrating.
TCAs, particularly in overdose, may cause ventricular dysrhythmias associated with prolongation of the QT interval (see Ch. 21). Usual therapeutic doses of TCAs increase, slightly but significantly, the risk of sudden cardiac death.
Interactions with other drugs
TCAs are particularly likely to cause adverse effects when given in conjunction with other drugs (see Ch. 56). They rely on hepatic metabolism by microsomal CYP enzymes for elimination, and this may be inhibited by competing drugs (e.g. antipsychotic drugs and some steroids).
TCAs potentiate the effects of alcohol and anaesthetic agents, for reasons that are not well understood, and deaths have occurred as a result of this, when severe respiratory depression has followed a bout of drinking. TCAs also interfere with the action of various antihypertensive drugs (see Ch. 22), with potentially dangerous consequences, so their use in hypertensive patients requires close monitoring.
Acute toxicity
TCAs are dangerous in overdose, and were at one time commonly used for suicide attempts, which was an important factor prompting the introduction of safer antidepressants. The main effects are on the central nervous system and the heart. The initial effect of TCA overdosage is to cause excitement and delirium, which may be accompanied by convulsions. This is followed by coma and respiratory depression lasting for some days. Atropine-like effects are pronounced, including dry mouth and skin, mydriasis and inhibition of gut and bladder. Anticholinesterase drugs have been used to counter atropine-like effects but are no longer recommended. Cardiac dysrhythmias (see above) are common, and sudden death (rare) may occur from ventricular fibrillation.
Pharmacokinetic aspects
TCAs are all rapidly absorbed when given orally and bind strongly to plasma albumin, most being 90–95% bound at therapeutic plasma concentrations. They also bind to extravascular tissues, which accounts for their generally very large distribution volumes (usually 10–50 l/kg; see Ch. 8) and low rates of elimination. Extravascular sequestration, together with strong binding to plasma albumin, means that haemodialysis is ineffective as a means of increasing drug elimination.
TCAs are metabolised in the liver by two main routes, N-demethylation, and ring hydroxylation (Fig. 46.4). Both the desmethyl and the hydroxylated metabolites commonly retain biological activity (see Table 46.4). During prolonged treatment with TCAs, the plasma concentration of these metabolites is usually comparable to that of the parent drug, although there is wide variation between individuals. Inactivation of the drugs occurs by glucuronide conjugation of the hydroxylated metabolites, the glucuronides being excreted in the urine.
The overall half-times for elimination of TCAs are generally long, ranging from 10–20 h for imipramine and desipramine to about 80 h for protriptyline. They are even longer in elderly patients. Therefore, gradual accumulation is possible, leading to slowly developing side effects. The relationship between plasma concentrations and the therapeutic effect is not simple. Indeed, a study on nortriptyline (Fig. 46.5) showed that too high a plasma concentration actually reduces the antidepressant effect, and there is a narrow ‘therapeutic window’.
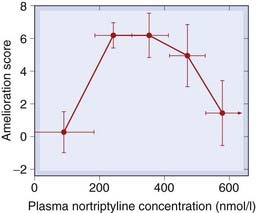
Fig. 46.5 ‘Therapeutic window’ for nortriptyline.
The antidepressant effect, determined from subjective rating scales, is optimal at plasma concentrations between 200 nmol/l and 400 nmol/l, and declines at higher levels.
Tricyclic antidepressants
Other Non-Selective Monoamine Uptake Inhibitors
Other relatively non-selective monoamine uptake inhibitors (often referred to as serotonin/noradrenaline reuptake inhibitors, or ‘SNRIs’) include venlafaxine, desvenlafaxine, duloxetine and milnacipran (see Table 46.2). These have become extensively used antidepressant drugs due to their perceived greater therapeutic efficacy and low side effect profiles.
Milnacipran has some selectivity for noradrenaline uptake over 5-HT uptake. As the dose of venlafaxine is increased, its efficacy also increases, which has been interpreted as demonstrating that its weak action to inhibit noradrenaline reuptake may add to its 5-HT uptake inhibition that occurs at lower doses, the combination providing additional therapeutic benefit. They are all active orally; venlafaxine is available in a slow-release formulation that reduces the incidence of nausea. Venlafaxine, desvenlafaxine and duloxetine are effective in some anxiety disorders (see Ch. 43). Desvenlafaxine may be useful in treating some perimenopausal symptoms such as hot flushes and insomnia. Duloxetine and milnacipran are used in the treatment of neuropathic pain and fibromyalgia (see Ch. 41). Duloxetine is also used to treat urinary incontinence.
Venlafaxine and duloxetine are metabolised by CYP2D6. Venlafaxine is converted to desvenlafaxine which shows greater inhibition of noradrenaline reuptake. Unwanted effects of these drugs—largely due to enhanced activation of adrenoceptors—include headache, insomnia, sexual dysfunction, dry mouth, dizziness, sweating and decreased appetite. The most common symptoms in overdose are CNS depression, serotonin toxicity, seizure and cardiac conduction abnormalities. Duloxetine has been reported to cause hepatotoxicity and is contraindicated for patients with hepatic impairment.
Other monoamine uptake inhibitors
Other Noradrenaline Uptake Inhibitors
Bupropion inhibits both noradrenaline and dopamine (but not 5-HT) uptake but, unlike cocaine and amphetamine (see Ch. 47), does not induce euphoria and has so far not been observed to have abuse potential. It is metabolised to active metabolites. It is also used to treat nicotine dependence (see Ch. 48). Reboxetine and atomoxetine are highly selective inhibitors of noradrenaline uptake but their efficacy in depression is less than that of TCAs. Atomoxetine is approved for the treatment of attention-deficit hyperactivity disorder (see Ch. 47).
Monoamine Receptor Antagonists
Mirtazapine blocks not only α2 adrenoreceptors but also other receptors, including 5-HT2C receptors, which may contribute to its antidepressant actions. Block of α2 adrenoceptors will not only increase noradrenaline release but will also enhance 5-HT release (see Fig 46.2B); however, by simultaneously blocking 5-HT2A and 5-HT3 receptors it will reduce unwanted effects mediated through these receptors (e.g. sexual dysfunction and nausea) but leave intact stimulation of postsynaptic 5-HT1A receptors. It also blocks histamine H1 receptors which may cause sedation. Trazodone combines 5-HT2A and 5-HT2C receptor antagonism with 5-HT reuptake inhibition.
Mianserin, another α2 adrenoceptor antagonist that also blocks H1, 5-HT2A and α1 adrenoreceptors, can cause bone marrow depression, requiring regular blood counts, so its use has declined in recent years.
Monoamine Oxidase Inhibitors
Monoamine oxidase inhibitors (MAOIs) were among the first drugs to be introduced clinically as antidepressants but were largely superseded by other types of antidepressants, whose clinical efficacies were considered better and whose side effects are generally less than those of MAOIs. The main examples are phenelzine, tranylcypromine and iproniazid. These drugs cause irreversible inhibition of the enzyme and do not distinguish between the two main isozymes. The discovery of reversible inhibitors that show isozyme selectivity has rekindled interest in this class of drug. Although several studies have shown a reduction in platelet MAO activity in certain groups of depressed patients, there is no clear evidence that abnormal MAO activity is involved in the pathogenesis of depression.
Monoamine oxidase (see Ch. 14) is found in nearly all tissues, and exists in two similar molecular forms coded by separate genes (see Table 46.5). MAO-A has a substrate preference for 5-HT and is the main target for the antidepressant MAOIs. MAO-B has a substrate preference for phenylethylamine and dopamine. Type B is selectively inhibited by selegiline, which is used in the treatment of Parkinson’s disease (see Ch. 39). Disruption of the MAO-A gene in mice causes increased brain accumulation of 5-HT and, to a lesser extent, noradrenaline, along with aggressive behaviour (Shih et al., 1999). A family has been reported with an inherited mutation leading to loss of MAO-A activity, whose members showed mental retardation and violent behaviour patterns. Most antidepressant MAOIs act on both forms of MAO, but clinical studies with subtype-specific inhibitors have shown clearly that antidepressant activity, as well as the main side effects of MAOIs, is associated with MAO-A inhibition. MAO is located intracellularly, mostly associated with mitochondria, and has two main functions:
Table 46.5 Substrates and inhibitors for type A and type B monoamine oxidase
| Type A | Type B | |
|---|---|---|
| Preferred substrates | Noradrenaline 5-Hydroxytryptamine |
Phenylethylamine Benzylamine |
| Non-specific substrates | Dopamine Tyramine |
Dopamine Tyramine |
| Specific inhibitors | Clorgyline Moclobemide |
Selegiline |
| Non-specific inhibitors | Pargyline Tranylcypromine Isocarboxazid |
Pargyline Tranylcypromine Isocarboxazid |
Chemical aspects
Monoamine oxidase inhibitors are substrate analogues with a phenylethylamine-like structure, and most contain a reactive group (e.g. hydrazine, propargylamine, cyclopropylamine) that enables the inhibitor to bind covalently to the enzyme, resulting in a non-competitive and long-lasting inhibition. Recovery of MAO activity after inhibition takes several weeks with most drugs, but is quicker after tranylcypromine, which forms a less stable bond with the enzyme. Moclobemide acts as a reversible competitive inhibitor.
Monoamine oxidase inhibitors are not particularly specific in their actions, and inhibit a variety of other enzymes as well as MAO, including many enzymes involved in the metabolism of other drugs. This is responsible for some of the many clinically important drug interactions associated with MAOIs.
Pharmacological effects
Monoamine oxidase inhibitors cause a rapid and sustained increase in the 5-HT, noradrenaline and dopamine content of the brain, 5-HT being affected most and dopamine least. Similar changes occur in peripheral tissues such as heart, liver and intestine, and increases in the plasma concentrations of these amines are also detectable. Although these increases in tissue amine content are largely due to accumulation within neurons, transmitter release in response to nerve activity is not increased. In contrast to the effect of TCAs, MAOIs do not increase the response of peripheral organs, such as the heart and blood vessels, to sympathetic nerve stimulation. The main effect of MAOIs is to increase the cytoplasmic concentration of monoamines in nerve terminals, without greatly affecting the vesicular stores that form the pool that is releasable by nerve stimulation. The increased cytoplasmic pool results in an increased rate of spontaneous leakage of monoamines, and also an increased release by indirectly acting sympathomimetic amines such as amphetamine and tyramine (see Ch. 14 and Fig. 14.8). Inhibition of MAO increases the proportion that escapes and thus enhances the response. Tyramine thus causes a much greater rise in blood pressure in MAOI-treated animals than in controls. This mechanism is important in relation to the cheese reaction produced by MAOIs in humans (see later section).
In normal human subjects, MAOIs cause an immediate increase in motor activity, and euphoria and excitement develop over the course of a few days. This is in contrast to TCAs, which cause only sedation and confusion when given to non-depressed subjects. The effects of MAOIs on amine metabolism develop rapidly, and the effect of a single dose lasts for several days. There is a clear discrepancy, as with SSRIs and TCAs, between the rapid biochemical response and the delayed antidepressant effect.
Unwanted effects and toxicity
Many of the unwanted effects of MAOIs result directly from MAO inhibition, but some are produced by other mechanisms.
Hypotension is a common side effect; indeed, pargyline was at one time used as an antihypertensive drug. One possible explanation for this effect—the opposite of what might have been expected—is that amines such as dopamine or octopamine accumulate within peripheral sympathetic nerve terminals and displace noradrenaline from the storage vesicles, thus reducing noradrenaline release associated with sympathetic activity.
Excessive central stimulation may cause tremors, excitement, insomnia and, in overdose, convulsions.
Increased appetite, leading to weight gain, can be so extreme as to require the drug to be discontinued.
Atropine-like side effects (dry mouth, blurred vision, urinary retention, etc.) are common with MAOIs, although they are less of a problem than with TCAs.
MAOIs of the hydrazine type (e.g. phenelzine and iproniazid) produce, very rarely (less than 1 in 10 000), severe hepatotoxicity, which seems to be due to the hydrazine moiety of the molecule. Their use in patients with liver disease is therefore unwise.
Interaction with other drugs and foods
Interaction with other drugs and foods is the most serious problem with MAOIs and is the main factor that caused their clinical use to decline. The special advantage claimed for the new reversible MAOIs, such as moclobemide, is that these interactions are reduced.
The cheese reaction is a direct consequence of MAO inhibition and occurs when normally innocuous amines (mainly tyramine) produced during fermentation are ingested. Tyramine is normally metabolised by MAO in the gut wall and liver, and little dietary tyramine reaches the systemic circulation. MAO inhibition allows tyramine to be absorbed, and also enhances its sympathomimetic effect, as discussed above. The result is acute hypertension, giving rise to a severe throbbing headache and occasionally even to intracranial haemorrhage. Although many foods contain some tyramine, it appears that at least 10 mg of tyramine needs to be ingested to produce such a response, and the main danger is from ripe cheeses and from concentrated yeast products such as Marmite. Administration of indirectly acting sympathomimetic amines (e.g. ephedrine—a nasal decongestant—or amphetamine—a drug of abuse) also causes severe hypertension in patients receiving MAOIs; directly acting agents such as noradrenaline (used, for example, in conjunction with local anaesthetics; see Ch. 42) are not hazardous. Moclobemide, a specific MAO-A inhibitor, does not cause the cheese reaction, probably because tyramine can still be metabolised by MAO-B.
Hypertensive episodes have been reported in patients given TCAs and MAOIs simultaneously. The probable explanation is that inhibition of noradrenaline reuptake further enhances the cardiovascular response to dietary tyramine, thus accentuating the cheese reaction. This combination of drugs can also produce excitement and hyperactivity.
Monoamine oxidase inhibitors can interact with pethidine (see Ch. 41) to cause severe hyperpyrexia, with restlessness, coma and hypotension. The mechanism is uncertain, but it is likely that an abnormal pethidine metabolite is produced because of inhibition of demethylation.
Other antidepressant drugs
Monoamine oxidase inhibitors (MAOIs)
Miscellaneous Agents
Methylfolate, given as a dietary supplement, may be effective in depressed individuals who have lowered folate levels.
Oestrogen which is known to elevate mood in perimenopausal women may also be of value for the treatment of postpartum depression. Its effectiveness in treating other forms of depression is unclear. In addition to its well documented hormonal actions in the body (see Ch. 34), it also has actions on monoaminergic, GABAergic and glutamatergic systems in the brain (see Chs 37 and 38).
Future Antidepressant Drugs
After a fallow period, there are now several promising new drugs in development (see Lodge & Li, 2008; Mathew et al., 2008). These can be classified broadly into the following:
Other avenues of research are into the development of compounds that act on the signal transduction pathways responsible for neurogenesis, neural plasticity and apoptosis.
Clinical uses of drugs in depression 
Brain Stimulation Therapies
There are now a number of brain stimulation techniques being used or developed to treat depression. The most established are electroconvulsive therapy (ECT) and repetitive transcranial magnetic stimulation (TMS). Brain stimulation treatments are often used as the therapeutic approach of last resort on patients who have not responded to antidepressant drugs.
ECT involves stimulation through electrodes placed on either side of the head, with the patient lightly anaesthetised, paralysed with a short-acting neuromuscular-blocking drug (e.g. suxamethonium; Ch. 13) so as to avoid physical injury, and artificially ventilated. Controlled trials have shown ECT to be at least as effective as antidepressant drugs, with response rates ranging between 60% and 80%; it appears to be an effective treatment for severe suicidal depression and has the advantage of producing a fast-onset response. The main disadvantage of ECT is that it often causes confusion and memory loss lasting for days or weeks. TMS gives electrical stimulation without anaesthesia or convulsion and does not produce cognitive impairment (see Kirkcaldie et al., 1997).
The effect of ECT on experimental animals has been carefully analysed to see if it provides clues as to the mode of action of antidepressant drugs, but the clues it gives are enigmatic. 5-HT synthesis and uptake are unaltered, and noradrenaline uptake is somewhat increased (in contrast to the effect of TCAs). Decreased β-adrenoceptor responsiveness, both biochemical and behavioural, occurs with both ECT and long-term administration of antidepressant drugs, but changes in 5-HT-mediated responses tend to go in opposite directions (see Maes & Meltzer, 1995).
There have been reports that deep brain stimulation, which has also been used in the treatment of Parkinson’s disease (see Ch. 39), in which the activity in a specific brain region is altered through surgically implanted electrodes, is effective in patients not responding to other treatments (see Mayberg et al., 2005). The effectiveness of another technique, vagal stimulation, in producing long-term benefit in depression is still unclear (see Grimm & Bajbouj, 2010).
Clinical Effectiveness of Antidepressant Treatments
The overall clinical efficacy of antidepressants has been established in many well-controlled clinical trials, although the degree of improvement may be limited. In long-term therapy, however, the remission rate can be as low as 30%. Moreover, it is clear that some patients recover spontaneously, and that 30–40% of patients fail to improve with drug treatments. Although antidepressants produce significant benefit in patients with moderate or severe depression, their efficacy in mild cases is unclear. Controlled trials show there is little to choose in terms of overall efficacy between any of the drugs currently in use, although clinical experience suggests that individual patients may, for unknown reasons, respond better to one drug than to another.
Pharmacogenetic factors
 The individual variation in response to antidepressants may be partly due to genetic factors, as well as to heterogeneity of the clinical condition. Two genetic factors have received particular attention, namely:
The individual variation in response to antidepressants may be partly due to genetic factors, as well as to heterogeneity of the clinical condition. Two genetic factors have received particular attention, namely:
Up to 10% of Caucasians possess a dysfunctional CYP2D6 gene, and consequently may be susceptible to side effects of TCAs and various other drugs (see Ch. 11) that are metabolised by this route. The opposite effect, caused by duplication of the gene, is common in Eastern European and East African populations, and may account for a lack of clinical efficacy in some individuals. There is some evidence to suggest that responsiveness to SSRIs is related to polymorphism of one of the serotonin transporter genes (see Gerretsen & Pollock, 2008).
Although genotyping may prove to be a useful approach in the future to individualising antidepressant therapy, its practical realisation is still some way off.
Suicide and antidepressants
Some years ago there were reports that antidepressants increased the risk of ‘suicidality’ in depressed patients, especially in children and adolescents (see Licinio & Wong, 2005). The term suicidality encompasses suicidal thoughts and planning as well as unsuccessful attempts; actual suicide, although one of the major causes of death in young people, is much rarer than suicidality. Clinical trials to determine the relationship between antidepressants and suicidality are difficult, because of the clear association between depression and suicide, and have given variable results, with some studies suggesting that suicidality may be increased during the first few weeks of antidepressant treatment, although not thereafter, and some showing a small increase in the risk of actual suicide (see Cipriani et al., 2005). Recent reviews of published data conclude that although antidepressants, including SSRIs, carry a small risk of inducing suicidal thoughts and suicide attempts in young people, the risk is less in older age groups (Hetrick et al., 2007; Möller et al., 2008; Barbui et al., 2009). There is no evidence to suggest that SSRIs carry any greater risk than other antidepressants. Furthermore, the risk has to be balanced against the beneficial effects of these drugs, not only on depression but also on anxiety, panic and obsessive-compulsive disorders (see Ch. 43).
Other Clinical Uses of Antidepressant Drugs
To some extent, the term ‘antidepressant drug’ is misleading as many of these drugs are now used to treat disorders other than depression. These include:
Drug Treatment of Bipolar Depression
A range of drugs are now used to control the mood swings characteristic of manic-depressive (bipolar) illness. The major drugs are:
When used to treat bipolar depression, lithium and antiepileptic agents are often referred to as mood-stabilising drugs.
Other agents that may have some beneficial effects in the treatment of bipolar depression are benzodiazepines (to calm, induce sleep and reduce anxiety), memantine, amantadine, and ketamine. The use of antidepressant drugs in bipolar depression is somewhat controversial. It is recommended that they are given in combination with an antimanic agent because, in some patients, they may induce or enhance mania.
Used prophylactically in bipolar depression, drugs prevent the swings of mood and thus can reduce both the depressive and the manic phases of the illness. They are given over long periods, and their beneficial effects take 3–4 weeks to develop. Given in an acute attack, they are effective only in reducing mania, but not the depressive phase (although lithium is sometimes used as an adjunct to antidepressants in severe cases of unipolar depression).
Lithium
The psychotropic effect of lithium was discovered in 1949 by Cade, who had predicted that urate salts should prevent the induction by uraemia of a hyperexcitability state in guinea pigs. He found lithium urate to produce an effect, quickly discovered that it was due to lithium rather than urate, and went on to show that lithium produced a rapid improvement in a group of manic patients.
Antiepileptic and atypical antipsychotic drugs (see below) are equally effective in treating acute mania; they act more quickly and are considerably safer, so the clinical use of lithium is mainly confined to prophylactic control of manic-depressive illness. The use of lithium is declining.5 It is relatively difficult to use, as plasma concentration monitoring is required, and there is the potential for problems in patients with renal impairment and for drug interactions, for example with diuretics (see Ch. 56). Lithium may have beneficial effects in neurodegenerative diseases such as Alzheimer’s disease (see Ch. 39).
Pharmacological effects and mechanism of action
Lithium is clinically effective at a plasma concentration of 0.5–1 mmol/l, and above 1.5 mmol/l it produces a variety of toxic effects, so the therapeutic window is narrow. In normal subjects, 1 mmol/l lithium in plasma has no appreciable psychotropic effects. It does, however, produce many detectable biochemical changes, and it is still unclear how these may be related to its therapeutic effect.
Lithium is a monovalent cation that can mimic the role of Na+ in excitable tissues, being able to permeate the voltage-gated Na+ channels that are responsible for action potential generation (see Ch. 4). It is, however, not pumped out by the Na+-K+-ATPase, and therefore tends to accumulate inside excitable cells, leading to a partial loss of intracellular K+, and depolarisation of the cell.
The biochemical effects of lithium are complex, and it inhibits many enzymes that participate in signal transduction pathways. Those that are thought to be relevant to its therapeutic actions are as follows:
Lithium also inhibits hormone-induced cAMP production and blocks other cellular responses (e.g. the response of renal tubular cells to antidiuretic hormone, and of the thyroid to thyroid-stimulating hormone; see Chs 28 and 33, respectively). This is not, however, a pronounced effect in the brain.
The cellular selectivity of lithium appears to depend on its selective uptake, reflecting the activity of sodium channels in different cells. This could explain its relatively selective action in the brain and kidney, even though many other tissues use the same second messengers. Notwithstanding such insights, our ignorance of the nature of the disturbance underlying the mood swings in bipolar depression leaves us groping for links between the biochemical and prophylactic effects of lithium.
Pharmacokinetic aspects and toxicity
Lithium is given by mouth as the carbonate salt and is excreted by the kidney. About half of an oral dose is excreted within about 12 h—the remainder, which presumably represents lithium taken up by cells, is excreted over the next 1–2 weeks. This very slow phase means that, with regular dosage, lithium accumulates slowly over 2 weeks or more before a steady state is reached. The narrow therapeutic window (approximately 0.5–1.5 mmol/l) means that monitoring of the plasma concentration is essential. Na+ depletion reduces the rate of excretion by increasing the reabsorption of lithium by the proximal tubule, and thus increases the likelihood of toxicity. Diuretics that act distal to the proximal tubule (Ch. 28) also have this effect, and renal disease also predisposes to lithium toxicity.
The main toxic effects that may occur during treatment are as follows:
Acute lithium toxicity results in various neurological effects, progressing from confusion and motor impairment to coma, convulsions and death if the plasma concentration reaches 3–5 mmol/l.
Antiepileptic Drugs
Carbamazepine, valproate and lamotrogine have fewer side effects than lithium and have proved efficacious in the treatment of bipolar depression.
It is assumed that the mechanisms of action of anticonvulsant drugs in reducing bipolar depression are the same as for their anticonvulsant activity. While each drug has multiple actions (see Table 44.1), the antiepileptic drugs effective in bipolar depression share the property of sodium channel blockade, although there are subtle differences in their effectiveness against the different phases of bipolar depression. Valproate and carbamazepine are effective in treating acute attacks of mania and in the long-term treatment of the disorder, although carbamazepine may not be as effective in treating the depression phase. Valproate is sometimes given along with other drugs such as lithium. Lamotrogine is effective in preventing the recurrence of both mania and depression. Riluzole, which was developed to treat amyotrophic lateral sclerosis (Ch. 39), has anticonvulsant activity in animal models. It may be useful in the treatment of bipolar disorders resistant to other agents.
The efficacy of gabapentin and pregabalin in bipolar depression has been questioned (see Stahl, 2008), but they may be useful as adjunct therapies to treat the chronic pain and anxiety that sufferers from bipolar depression may also experience. Levetiracetam, topiramate and zonisamide are sometimes used in the treatment of bipolar depression but their efficacy still remains to be established.
Atypical Antipsychotic Drugs
Atypical antipsychotic drugs (e.g. olanzapine, risperidone, quetiapine, aripiprazole) are second-generation drugs developed for the treatment of schizophrenia (see Ch. 45). These agents have D2 dopamine and 5-HT2A receptor antagonist properties as well as actions on other receptors and amine transporters that may contribute to their effectiveness in bipolar depression. All appear to be effective against mania while some may also be effective against bipolar depression. In bipolar depression, atypical antipsychotics are often used in combination with lithium or valproate. Olanzepine is given in combination with the antidepressant fluoxetine.
Treatment of bipolar depression
Clinical uses of mood-stabilising drugs
References and Further Reading
Pathogenesis of depressive illness
Charney D.S., Manji M.K. Life stress, genes and depression: multiple pathways lead to increased risk and new opportunities for intervention. Sci. STKE.. 2004. 2004: re5 http://www.stke.org (Detailed review of current understanding of the pathophysiology of depression, emphasising the role of neural plasticity, neurogenesis and apoptosis)
Cryan J.F., Slattery D.A. Animal models of mood disorders: recent developments. Curr. Opin. Psychiatry. 2007;20:1-7. (Useful review of animal models)
Duman R.S. Depression: a case of neuronal life and death? Biol. Psychiatry. 2004;56:140-145. (Reviews evidence suggesting that neuronal loss in the hippocampus and prefrontal cortex results in depressive symptoms, and that antidepressants act indirectly to promote neurogenesis)
Gardier A.M. Mutant mouse models and antidepressant drug research: focus on serotonin and brain-derived neurotrophic factor. Behav. Pharmacol.. 2009;20:18-32.
Maes M., Meltzer H.Y. The serotonin hypothesis of major depression. In: Bloom F.E., Kupfer D.J., editors. Psychopharmacology: the fourth generation of progress. New York: Raven Press, 1995. (Review showing how emphasis has shifted towards the involvement of 5-HT, rather than noradrenaline, in the aetiology of depression)
Manji H.K., Drevets W.C., Charney D.S. The cellular neurobiology of depression. Nat. Med.. 2001;7:541-547. (Speculative review of the possible mechanisms and role of neurodegeneration and neuroplasticity in depressive disorders, attempting to move beyond the monoamine theory)
Santarelli L., Saxe M., Gross C., et al. Requirement of hippocampal neurogenesis for the behavioural effects of antidepressants. Science. 2003;301:805-809. (Study in rats suggesting that growth of new hippocampal neurons is responsible for antidepressant effects; commentary in same issue, p. 757)
Shih J.C., Chen K., Ridd M.J. Monoamine oxidase: from genes to behaviour. Annu. Rev. Neurosci.. 1999;22:197-217. (Review of recent work on transgenic mice with MAO mutation or deletion)
Ballasteros J., Callado L.F. Effectiveness of pindolol plus serotonin uptake inhibitors in depression: a meta-analysis of early and late outcomes from randomised controlled trials. J. Affect. Disord.. 2004;79:137-147.
Barbui C., Esposito E., Cipriani A. Selective serotonin reuptake inhibitors and risk of suicide: a systematic review of observational studies. Can. Med. Assoc. J.. 2009;180:291-297.
Bosker F.J., Westerink B.H., Cremers T.I., et al. Future antidepressants: what is in the pipeline and what is missing? CNS Drugs. 2004;18:705-732. (Focuses on new approaches to development of antidepressants)
Cipriani A., Barbui C., Geddes J.R. Suicide, depression, and antidepressants. Br. Med. J.. 2005;330:373-374. (Comment on detailed trials data in the same issue of the journal)
Cipriani, A., Santilli, C., Furukawa, T.A., et al., 2009. Escitalopram versus other antidepressive agents for depression. Cochrane Database Syst. Rev., Issue 2. Art. No.: CD006532. DOI: 10.1002/14651858.CD006532.pub2.
Gerretsen P., Pollock B.G. Pharmacogenetics and the serotonin transporter in late-life depression. Exp. Opin. Drug Metab. Toxicol.. 2008;4:1465-1478.
Glatt C.E., Reus V.I. Pharmacogenetics of monoamine transporters. Pharmacogenomics. 2003;4:583-596. (Discusses prospects for correlating transporter gene polymorphism to variation in response to psychoactive drugs)
Grimm S., Bajbouj M. Efficacy of vagus nerve stimulation in the treatment of depression. Expert. Rev. Neurother.. 2010;10:87-92.
Hetrick, S.E., Merry, S.N., McKenzie, J., Sindahl, P., Proctor, M., 2007. Selective serotonin reuptake inhibitors (SSRIs) for depressive disorders in children and adolescents. Cochrane Database Syst. Rev., Issue 3. Art. No.: CD004851. DOI: 10.1002/14651858.CD004851.pub2.
Holsboer F. The rationale for corticotrophin-releasing hormone receptor (CRH-R) antagonists to treat depression and anxiety. J. Psychiatr. Res.. 1999;33:181-214. (Reviews the evidence linking CRH with depressive illness)
Kirchheiner J., Nickchen K., Bauer M., et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol. Psychiatry. 2004;9:442-473. (Discusses effect of gene polymorphisms on antidepressant actions; principles are not yet incorporated into clinical practice)
Kirkcaldie M.T., Pridmore S.A., Pascual-Leone A. Transcranial magnetic stimulation as therapy for depression and other disorders. Aust. N. Z. J. Psychiatry. 1997;31:264-272.
Licinio J., Wong M.-L. Depression, antidepressants and suicidality: a critical appraisal. Nat. Rev. Drug Discov.. 2005;4:165-171. (Review of the equivocal evidence linking antidepressant use to suicide)
Lodge N.J., Li Y.-W. Ion channels as potential targets for the treatment of depression. Curr. Opin. Drug Discov. Devel.. 2008;11:633-641.
Mathew S.J., Manji H.K., Charney D.S. Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology. 2008;33:2080-2092.
Mayberg H.S., Lozano A.M., Voon V., et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651-660.
Möller H.-J., Baldwin D.S., Goodwin G., et al. Do SSRIs or antidepressants in general increase suicidality? J. Clin. Invest.. 2008;119:717-725.
Pacher P., Kecsemeti V. Trends in the development of new antidepressants. Is there light at the end of the tunnel? Curr. Med. Chem.. 2004;11:925-943. (Discusses new developments from the starting point of the monoamine theory)
Racagni G., Popoli M. Cellular and molecular mechanisms in the long-term action of antidepressants. Dialogues. Clin. Neurosci.. 2008;10:385-400. (An extensive review of long-term changes induced in the brain by antidepressant drugs that may be responsible for producing the therapeutic benefit)
Song F., Freemantle N., Sheldon T.A., et al. Selective serotonin reuptake inhibitors: meta-analysis of efficacy and acceptability. Br. Med. J.. 1993;306:683-687. (Summary of clinical trials data, showing limitations as well as advantages of SSRIs)
Beaulieu J.M., Gainetdinov R.R., Caron M.G. Akt/GSK3 signaling in the action of psychotropic drugs. Annu. Rev. Pharmacol. Toxicol.. 2009;49:327-347.
Phiel C.J., Klein P.S. Molecular targets of lithium action. Annu. Rev. Pharmacol. Toxicol.. 2001;41:789-813. (Review of a topic that is still little understood)
1Neurogenesis (see Ch. 39)—the formation of new neurons from stem cell precursors—occurs to a significant degree in the adult hippocampus, and possibly elsewhere in the brain, contradicting the old dogma that it occurs only during brain development.
2Cynics may feel that these mechanisms, in which glutamate, neurotrophic factors, monoamines and steroids all interact to control neuronal death, survival and plasticity, are being invoked just as enthusiastically to account for almost every neurological and psychiatric disorder that you can think of, from stroke and Parkinson’s disease to schizophrenia. ‘Are we missing something,’ they may feel, ‘or are all these diseases basically the same? If so, why are their effects so different? Is this just a scientific bandwagon, or does this mechanistic convergence point to some fundamental principles of neural organisation?’ We do not have the answers, of course, but it is a field worth watching.
3Although relatively free of acute side effects, hyperforin activates cytochrome P450, resulting in loss of efficacy, with serious consequences, of several important drugs, including ciclosporin, oral contraceptives, some anti-HIV and anticancer drugs, and oral anticoagulants—underlining the principle that herbal remedies need to be used with the same degree of informed caution as any other drug.
4Thus, conversely, SSRIs can be used to treat premature ejaculation.
5The decline in lithium use may have been influenced by the imbalance in the marketing of this simple inorganic ion versus other pharmacological agents.