13 Cholinergic transmission
Overview
This chapter is concerned mainly with cholinergic transmission in the periphery, and the ways in which drugs affect it. Here we describe the different types of acetylcholine (ACh) receptors and their functions, as well as the synthesis and release of ACh. Drugs that act on ACh receptors, many of which have clinical uses, are described in this chapter. Cholinergic mechanisms in the central nervous system (CNS) and their relevance to dementia are discussed in Chapters 38 and 39.
Muscarinic and Nicotinic Actions of Acetylcholine
 The discovery of the pharmacological action of ACh came, paradoxically, from work on adrenal glands, extracts of which were known to produce a rise in blood pressure owing to their content of adrenaline (epinephrine). In 1900, Reid Hunt found that after adrenaline had been removed from such extracts, they produced a fall in blood pressure instead of a rise. He attributed the fall to the presence of choline, but later concluded that a more potent derivative of choline must be responsible. With Taveau, he tested a number of choline derivatives and discovered that ACh was some 100 000 times more active than choline in lowering the rabbit’s blood pressure. The physiological role of ACh was not apparent at that time, and it remained a pharmacological curiosity until Loewi and Dale and their colleagues discovered its transmitter role in the 1930s.
The discovery of the pharmacological action of ACh came, paradoxically, from work on adrenal glands, extracts of which were known to produce a rise in blood pressure owing to their content of adrenaline (epinephrine). In 1900, Reid Hunt found that after adrenaline had been removed from such extracts, they produced a fall in blood pressure instead of a rise. He attributed the fall to the presence of choline, but later concluded that a more potent derivative of choline must be responsible. With Taveau, he tested a number of choline derivatives and discovered that ACh was some 100 000 times more active than choline in lowering the rabbit’s blood pressure. The physiological role of ACh was not apparent at that time, and it remained a pharmacological curiosity until Loewi and Dale and their colleagues discovered its transmitter role in the 1930s.
Analysing the pharmacological actions of ACh in 1914, Dale distinguished two types of activity, which he designated as muscarinic and nicotinic because they mimicked, respectively, the effects of injecting muscarine, the active principle of the poisonous mushroom Amanita muscaria, and of injecting nicotine. Muscarinic actions closely resemble the effects of parasympathetic stimulation, as shown in Table 12.1. After the muscarinic effects have been blocked by atropine, larger doses of ACh produce nicotine-like effects, which include:
The muscarinic and nicotinic actions of ACh are demonstrated in Figure 13.1. Small and medium doses of ACh produce a transient fall in blood pressure due to arteriolar vasodilatation and slowing of the heart—muscarinic effects that are abolished by atropine. A large dose of ACh given after atropine produces nicotinic effects: an initial rise in blood pressure due to a stimulation of sympathetic ganglia and consequent vasoconstriction, and a secondary rise resulting from secretion of adrenaline.
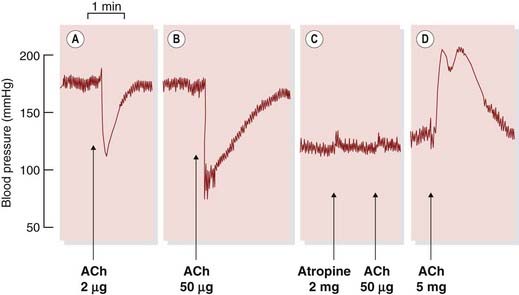
Fig. 13.1 Dale’s experiment showing that acetylcholine (ACh) produces two kinds of effect on the cat’s blood pressure.
Arterial pressure was recorded with a mercury manometer from a spinal cat. [A] ACh causes a fall in blood pressure due to vasodilatation. [B] A larger dose also produces bradycardia. Both [A] and [B] are muscarinic effects. [C] After atropine (muscarinic antagonist), the same dose of ACh has no effect. [D] Still under the influence of atropine, a much larger dose of ACh causes a rise in blood pressure (due to stimulation of sympathetic ganglia), accompanied by tachycardia, followed by a secondary rise (due to release of adrenaline from the adrenal gland). These effects result from its action on nicotinic receptors.
(From Burn J H 1963 Autonomic pharmacology. Blackwell, Oxford.)
Dale’s pharmacological classification corresponds closely to the main physiological functions of ACh in the body. The muscarinic actions correspond to those of ACh released at postganglionic parasympathetic nerve endings, with two significant exceptions:
The nicotinic actions correspond to those of ACh acting on autonomic ganglia of the sympathetic and parasympathetic systems, the motor endplate of voluntary muscle and the secretory cells of the adrenal medulla.
Acetylcholine Receptors
Although Dale himself dismissed the concept of receptors as sophistry rather than science, his functional classification provided the basis for distinguishing the two major classes of ACh receptor (see Ch. 3).
Nicotinic Receptors
Nicotinic ACh receptors (nAChRs) fall into three main classes, the muscle, ganglionic and CNS types, whose subunit compositions are summarised in Table 13.1. Muscle receptors are confined to the skeletal neuromuscular junction; ganglionic receptors are responsible for transmission at sympathetic and parasympathetic ganglia; and CNS-type receptors are widespread in the brain, and are heterogeneous with respect to their molecular composition and location (see Ch. 38).
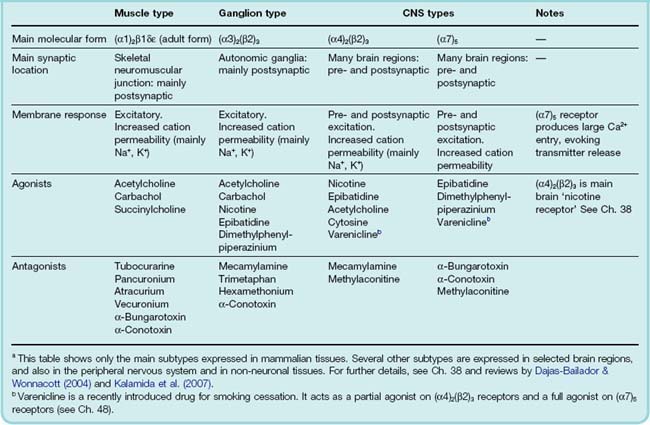
All nAChRs are pentameric structures that function as ligand-gated ion channels (see Ch. 3). The five subunits that form the receptor–channel complex are similar in structure, and so far 17 different members of the family have been identified and cloned, designated α (10 types), β (4 types), γ, δ and ε (one of each). The five subunits each possess four membrane-spanning helical domains, and one of these helices (M2) from each subunit defines the central pore (see Ch. 3). nAChR subtypes generally contain both α and β subunits, the exception being the homomeric (α7)5 subtype found mainly in the brain (Ch. 38). The adult muscle receptor has the composition (α1)2/β1εδ, while the main ganglionic subtype is (α3)2(β2)3. The two binding sites for ACh (both of which need to be occupied to cause the channel to open) reside at the interface between the extracellular domain of each of the α subunits and its neighbour. The diversity of the nAChR family (for details see Hogg et al., 2003 and Kalamida et al., 2007), which emerged from cloning studies in the 1980s, took pharmacologists somewhat by surprise. Although they knew that the neuromuscular and ganglionic synapses differed pharmacologically, and suspected that cholinergic synapses in the CNS might be different again, the molecular diversity goes far beyond this, and its functional significance is only slowly emerging.
The different action of agonists and antagonists on neuromuscular, ganglionic and brain synapses is of practical importance and mainly reflects the differences between the muscle and neuronal nAChRs (Table 13.1).
Muscarinic Receptors
Muscarinic receptors (mAChRs) are typical G-protein-coupled receptors (see Ch. 3), and five molecular subtypes (M1–M5) are known (see Wess, 1996). The odd-numbered members of the group (M1, M3, M5) couple with Gq to activate the inositol phosphate pathway (Ch. 3), while the even-numbered receptors (M2, M4) act through Gi to inhibit adenylyl cyclase and thus reduce intracellular cAMP (see Goyal, 1989). Both groups activate the MAP kinase pathway. The location and pharmacology of these subtypes are summarized in Table 13.2.
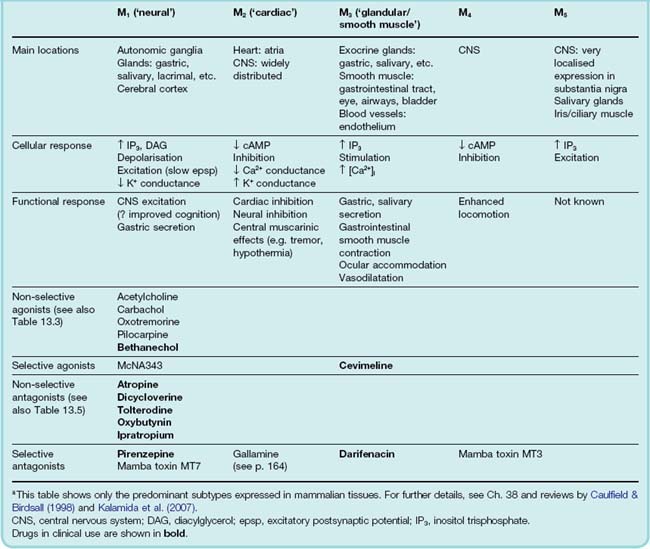
M1 receptors (‘neural’) are found mainly on CNS and peripheral neurons and on gastric parietal cells. They mediate excitatory effects, for example the slow muscarinic excitation mediated by ACh in sympathetic ganglia (Ch. 12) and central neurons. This excitation is produced by a decrease in K+ conductance, which causes membrane depolarisation. Deficiency of this kind of ACh-mediated effect in the brain is possibly associated with dementia (see Ch. 39), although transgenic M1 receptor knockout mice show only slight cognitive impairment (see Wess et al., 2007). M1 receptors are also involved in the increase of gastric acid secretion following vagal stimulation (see Ch. 29).
M2 receptors (‘cardiac’) occur in the heart, and also on the presynaptic terminals of peripheral and central neurons. They exert inhibitory effects, mainly by increasing K+ conductance and by inhibiting calcium channels (see Ch. 4). M2 receptor activation is responsible for cholinergic inhibition of the heart, as well as presynaptic inhibition in the CNS and periphery (Ch. 12). They are also co-expressed with M3 receptors in visceral smooth muscle, and contribute to the smooth-muscle-stimulating effect of muscarinic agonists in several organs.
M3 receptors (‘glandular/smooth muscle’) produce mainly excitatory effects, i.e. stimulation of glandular secretions (salivary, bronchial, sweat, etc.) and contraction of visceral smooth muscle. M3 receptors also mediate relaxation of smooth muscle (mainly vascular), which results from the release of nitric oxide from neighbouring endothelial cells (Ch. 20). M1, M2 and M3 receptors occur also in specific locations in the CNS (see Ch. 38).
M4 and M5 receptors are largely confined to the CNS, and their functional role is not well understood, although mice lacking these receptors do show behavioural changes (Wess et al., 2007). Recently it has been discovered that cytokine secretion from lymphocytes and other cells is regulated by M1 and M3 receptors, while M2 and M4 receptors affect cell proliferation in various situations, opening up hitherto unsuspected therapeutic roles for muscarine receptor ligands (see Wessler & Kirkpatrick, 2008).
The agonist binding region is highly conserved between the different subtypes, so attempts to develop selective agonists and antagonists have had limited success. Most known agonists are non-selective, though two experimental compounds, McNA343 and oxotremorine, are selective for M1 receptors, on which carbachol is relatively inactive. Other M1 selective agents (e.g. xanomeline) are in development as possible treatments for dementia.1
There is more selectivity among antagonists. Although most of the classic muscarinic antagonists (e.g. atropine, hyoscine) are non-selective, pirenzepine is selective for M1 receptors, and darifenacin for M2 receptors. Gallamine, better known as a neuromuscular-blocking drug (see p. 164), is also a selective, although weak, M2 receptor antagonist.2 Recently, toxins from the venom of the green mamba have been discovered to be highly selective mAChR antagonists (see Table 13.2), as well as various synthetic compounds with some degree of selectivity (see Eglen et al., 1999, for more details). Compounds that have recently been approved for clinical use are described below (p. 160).
Acetylcholine receptors 
Physiology of Cholinergic Transmission
The physiology of cholinergic neurotransmission3 is described in detail by Nicholls et al. (2001). The main ways in which drugs can affect cholinergic transmission are shown in Figure 13.2.
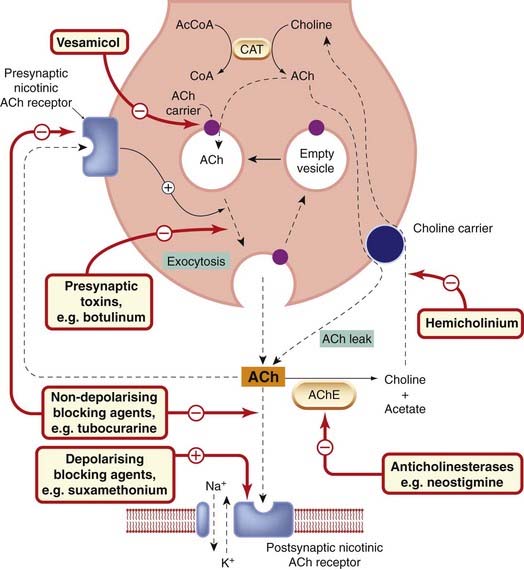
Fig. 13.2 Events and sites of drug action at a nicotinic cholinergic synapse.
Acetylcholine (ACh) is shown acting postsynaptically on a nicotinic receptor controlling a cation channel (e.g. at the neuromuscular or ganglionic synapse), and also on a presynaptic nicotinic receptor that acts to facilitate ACh release during sustained synaptic activity. The nerve terminal also contains acetylcholinesterase (not shown); when this is inhibited, the amount of free ACh, and the rate of leakage of ACh via the choline carrier, is increased. Under normal conditions, this leakage of ACh is insignificant. At muscarinic cholinergic junctions (e.g. heart, smooth muscle and exocrine glands), both postsynaptic and presynaptic (inhibitory) receptors are of the muscarinic type. AcCoA, acetyl coenzyme A; AChE, acetylcholinesterase; CAT, choline acetyltransferase; CoA, coenzyme A.
Acetylcholine Synthesis and Release
Acetylcholine metabolism is well reviewed by Parsons et al. (1993). ACh is synthesised within the nerve terminal from choline, which is taken up into the nerve terminal by a specific transporter (Ch. 12), similar to that which operates for many transmitters. The difference is that it transports the precursor, choline, not ACh, so it is not important in terminating the action of the transmitter. The concentration of choline in the blood and body fluids is normally about 10 µmol/l, but in the immediate vicinity of cholinergic nerve terminals it increases, probably to about 1 mmol/l, when the released ACh is hydrolysed, and more than 50% of this choline is normally recaptured by the nerve terminals. Free choline within the nerve terminal is acetylated by a cytosolic enzyme, choline acetyltransferase (CAT), which transfers the acetyl group from acetyl coenzyme A. The rate-limiting process in ACh synthesis appears to be choline transport, the activity of which is regulated according to the rate at which ACh is being released. Cholinesterase is present in the presynaptic nerve terminals, and ACh is continually being hydrolysed and resynthesised. Inhibition of the nerve terminal cholinesterase causes the accumulation of ‘surplus’ ACh in the cytosol, which is not available for release by nerve impulses (although it is able to leak out via the choline carrier). Most of the ACh synthesised, however, is packaged into synaptic vesicles, in which its concentration is very high (about 100 mmol/l), and from which release occurs by exocytosis triggered by Ca2+ entry into the nerve terminal (see Ch. 4).
Cholinergic vesicles accumulate ACh actively, by means of a specific transporter (see Usdin et al., 1995) belonging to the family of amine transporters described in Chapter 12. Accumulation of ACh is coupled to the large electrochemical gradient for protons that exists between intracellular organelles and the cytosol; it is blocked selectively by the experimental drug vesamicol (see Parsons et al., 1993). Following its release, the ACh diffuses across the synaptic cleft to combine with receptors on the postsynaptic cell. Some of it succumbs on the way to hydrolysis by acetylcholinesterase (AChE), an enzyme that is bound to the basement membrane, which lies between the pre- and postsynaptic membranes. At fast cholinergic synapses (e.g. the neuromuscular and ganglionic synapses), but not at slow ones (smooth muscle, gland cells, heart, etc.), the released ACh is hydrolysed very rapidly (within 1 ms), so that it acts only very briefly.
At the neuromuscular junction, which is a highly specialised synapse, a single nerve impulse releases about 300 synaptic vesicles (altogether about three million ACh molecules) from the nerve terminals supplying a single muscle fibre, which contain altogether about three million synaptic vesicles. Approximately two million ACh molecules combine with receptors, of which there are about 30 million on each muscle fibre, the rest being hydrolysed without reaching a receptor. The ACh molecules remain bound to receptors for, on average, about 2 ms, and are quickly hydrolysed after dissociating, so that they cannot combine with a second receptor. The result is that transmitter action is very rapid and very brief, which is important for a synapse that has to initiate speedy muscular responses, and that may have to transmit signals faithfully at high frequency. Muscle cells are much larger than neurons and require much more synaptic current to generate an action potential. Thus all the chemical events happen on a larger scale than at a neuronal synapse; the number of transmitter molecules in a quantum, the number of quanta released, and the number of receptors activated by each quantum are all 10–100 times greater. Our brains would be huge, but not very clever, if their synapses were built on the industrial scale of the neuromuscular junction.
Presynaptic Modulation
Acetylcholine release is regulated by mediators, including ACh itself, acting on presynaptic receptors, as discussed in Chapter 12. At postganglionic parasympathetic nerve endings, inhibitory M2 receptors participate in autoinhibition of ACh release; other mediators, such as noradrenaline, also inhibit the release of ACh (see Ch. 12). At the neuromuscular junction, on the other hand, presynaptic nAChRs facilitate ACh release (see Prior et al., 1995), a mechanism that may allow the synapse to function reliably during prolonged high-frequency activity. In the brain (see review by Dajas-Bailador & Wonnacott, 2004), most of the nAChRs are located presynaptically and serve to facilitate or inhibit the release of other mediators, such as glutamate and dopamine.
Electrical Events in Transmission at Fast Cholinergic Synapses
Acetylcholine, acting on the postsynaptic membrane of a nicotinic (neuromuscular or ganglionic) synapse, causes a large increase in its permeability to cations, particularly to Na+ and K+, and to a lesser extent Ca2+. The resulting inflow of Na+ depolarises the postsynaptic membrane. This transmitter-mediated depolarisation is called an endplate potential (epp) in a skeletal muscle fibre, or a fast excitatory postsynaptic potential (fast epsp) at the ganglionic synapse. In a muscle fibre, the localised epp spreads to adjacent, electrically excitable parts of the muscle fibre; if its amplitude reaches the threshold for excitation, an action potential is initiated, which propagates to the rest of the fibre and evokes a contraction (Ch. 4).
In a nerve cell, depolarisation of the soma or a dendrite by the fast epsp causes a local current to flow. This depolarises the axon hillock region of the cell, where, if the epsp is large enough, an action potential is initiated. Figure 13.3 shows that tubocurarine, a drug that blocks postsynaptic ACh receptors (see p. 164), reduces the amplitude of the fast epsp until it no longer initiates an action potential, although the cell is still capable of responding when it is stimulated antidromically. Most ganglion cells are supplied by several presynaptic axons, and it requires simultaneous activity in more than one to make the postganglionic cell fire. At the neuromuscular junction, only one nerve fibre supplies each muscle fibre. Nevertheless, the amplitude of the epp is normally more than enough to initiate an action potential—indeed, transmission still occurs when the epp is reduced by 70–80%, and is said to show a large margin of safety so that fluctuations in transmitter release (e.g. during repetitive stimulation) do not affect transmission.
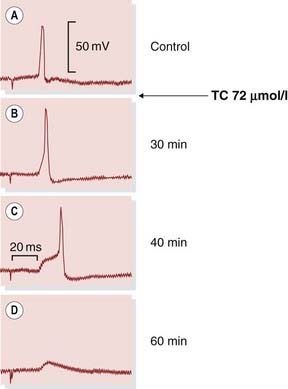
Fig. 13.3 Cholinergic transmission in an autonomic ganglion cell.
Records were obtained with an intracellular microelectrode from a guinea pig parasympathetic ganglion cell. The artefact at the beginning of each trace shows the moment of stimulation of the preganglionic nerve. Tubocurarine (TC), an acetylcholine antagonist, causes the epsp to become smaller. In record [C], it only just succeeds in triggering the action potential, and in [D] it has fallen below the threshold. Following complete block, antidromic stimulation (not shown) will still produce an action potential (cf. depolarisation block, Fig. 13.4).
(From Blackman J G et al. 1969 J Physiol 201: 723.)
Transmission at the ganglionic synapse is more complex than at the neuromuscular junction. Although the primary event at both is the epp or fast epsp produced by ACh acting on nAChRs, this is followed in the ganglion by a succession of much slower postsynaptic responses, comprising the following:
Cholinergic transmission
Depolarisation Block
Depolarisation block occurs at cholinergic synapses when the excitatory nAChRs are persistently activated, and it results from a decrease in the electrical excitability of the postsynaptic cell. This is shown in Figure 13.4. Application of nicotine to a sympathetic ganglion causes a depolarisation of the cell, which at first initiates action potential discharge. After a few seconds, this discharge ceases and transmission is blocked. The loss of electrical excitability at this time is shown by the fact that antidromic stimuli also fail to produce an action potential. The main reason for the loss of electrical excitability during a period of maintained depolarisation is that the voltage-sensitive sodium channels (see Ch. 4) become inactivated (i.e. refractory) and no longer able to open in response to a brief depolarising stimulus.
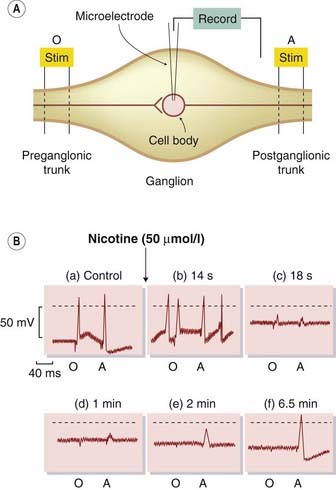
Fig. 13.4 Depolarisation block of ganglionic transmission by nicotine.
[A] System used for intracellular recording from sympathetic ganglion cells of the frog, showing the location of orthodromic (O) and antidromic (A) stimulating (stim) electrodes. Stimulation at O excites the cell via the cholinergic synapse, whereas stimulation at A excites it by electrical propagation of the action potential. [B] The effect of nicotine. (a) Control records. The membrane potential is −55 mV (dotted line = 0 mV), and the cell responds to both O and A. (b) Shortly after adding nicotine, the cell is slightly depolarised and spontaneously active, but still responsive to O and A. (c and d) The cell is further depolarised, to −25 mV, and produces only a vestigial action potential. The fact that it does not respond to A shows that it is electrically inexcitable. (e and f) In the continued presence of nicotine, the cell repolarises and regains its responsiveness to A, but it is still unresponsive to O because the ACh receptors are desensitised by nicotine.
(From Ginsborg B L, Guerrero S 1964 J Physiol 172: 189.)
A second type of effect is also seen in the experiment shown in Figure 13.4. After nicotine has acted for several minutes, the cell partially repolarises and its electrical excitability returns but, despite this, transmission remains blocked. This type of secondary, non-depolarising block occurs also at the neuromuscular junction if repeated doses of the depolarising drug suxamethonium4 (see below) are used. The main factor responsible for the secondary block (known clinically as phase II block) appears to be receptor desensitisation (see Ch. 2). This causes the depolarising action of the blocking drug to subside, but transmission remains blocked because the receptors are desensitised to ACh.
Effects of Drugs on Cholinergic Transmission
As shown in Figure 13.2, drugs can influence cholinergic transmission either by acting on postsynaptic ACh receptors as agonists or antagonists (Tables 13.1 and 13.2), or by affecting the release or destruction of endogenous ACh.
In the rest of this chapter, we describe the following groups of drugs, subdivided according to their site of action:
Drugs Affecting Muscarinic Receptors
Muscarinic Agonists
Structure–activity relationships
Muscarinic agonists, as a group, are often referred to as parasympathomimetic, because the main effects that they produce in the whole animal resemble those of parasympathetic stimulation. The structures of acetylcholine and related choline esters are given in Table 13.3. They are agonists at both mAChRs and nAChRs, but act more potently on mAChRs (see Fig. 13.1). Bethanechol, pilocarpine and cevimeline (a recent introduction) are the only ones used clinically.
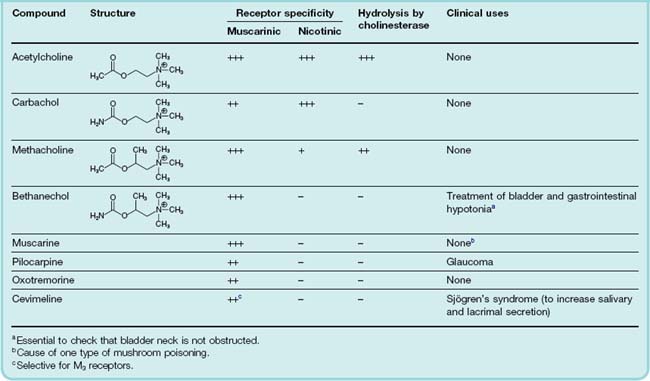
The key features of the ACh molecule that are important for its activity are the quaternary ammonium group, which bears a positive charge, and the ester group, which bears a partial negative charge and is susceptible to rapid hydrolysis by cholinesterase. Variants of the choline ester structure (Table 13.3) have the effect of reducing the susceptibility of the compound to hydrolysis by cholinesterase, and altering the relative activity on mAChRs and nAChRs.
Carbachol and methacholine are used as experimental tools. Bethanechol, which is a hybrid of these two molecules, is stable to hydrolysis and selective for mAChRs, and is occasionally used clinically. Pilocarpine is a partial agonist and shows some selectivity in stimulating secretion from sweat, salivary, lacrimal and bronchial glands, and contracting iris smooth muscle (see below), with weak effects on gastrointestinal smooth muscle and the heart.
Effects of muscarinic agonists
The main actions of muscarinic agonists are readily understood in terms of the parasympathetic nervous system.
Cardiovascular effects
These include cardiac slowing and a decrease in cardiac output. The latter action is due mainly to a decreased force of contraction of the atria, because the ventricles have only a sparse parasympathetic innervation and a low sensitivity to muscarinic agonists. Generalised vasodilatation also occurs (a nitric oxide-mediated effect; see Ch. 20), and these two effects combine to produce a sharp fall in arterial pressure (Fig. 13.1). The mechanism of action of muscarinic agonists on the heart is discussed in Chapter 21.
Smooth muscle
Smooth muscle other than vascular smooth muscle contracts in response to muscarinic agonists. Peristaltic activity of the gastrointestinal tract is increased, which can cause colicky pain, and the bladder and bronchial smooth muscle also contract.
Sweating, lacrimation, salivation and bronchial secretion
These result from stimulation of exocrine glands. The combined effect of bronchial secretion and constriction can interfere with breathing.
Effects on the eye
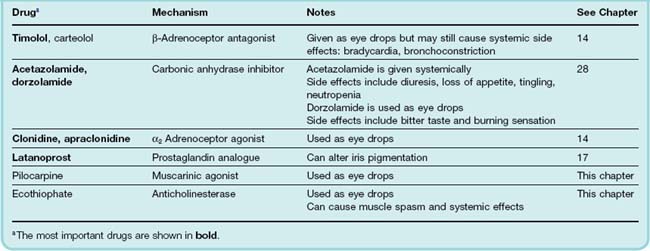
Such effects are of some importance. The parasympathetic nerves to the eye supply the constrictor pupillae muscle, which runs circumferentially in the iris, and the ciliary muscle, which adjusts the curvature of the lens (Fig. 13.5). Contraction of the ciliary muscle in response to activation of mAChRs pulls the ciliary body forward and inward, thus relaxing the tension on the suspensory ligament of the lens, allowing the lens to bulge more and reducing its focal length. This parasympathetic reflex is thus necessary to accommodate the eye for near vision. The constrictor pupillae is important not only for adjusting the pupil in response to changes in light intensity, but also in regulating the intraocular pressure. Aqueous humour is secreted slowly and continuously by the cells of the epithelium covering the ciliary body, and it drains into the canal of Schlemm (Fig. 13.5), which runs around the eye close to the outer margin of the iris. The intraocular pressure is normally 10–15 mmHg above atmospheric, which keeps the eye slightly distended. Abnormally raised intraocular pressure (associated with glaucoma) damages the eye and is one of the commonest preventable causes of blindness. In acute glaucoma, drainage of aqueous humour becomes impeded when the pupil is dilated, because folding of the iris tissue occludes the drainage angle, causing the intraocular pressure to rise. Activation of the constrictor pupillae muscle by muscarinic agonists in these circumstances lowers the intraocular pressure, although in a normal individual it has little effect. The increased tension in the ciliary muscle produced by these drugs may also play a part in improving drainage by realigning the connective tissue trabeculae through which the canal of Schlemm passes. Drugs used in the treatment of glaucoma are summarised in Table 13.4.
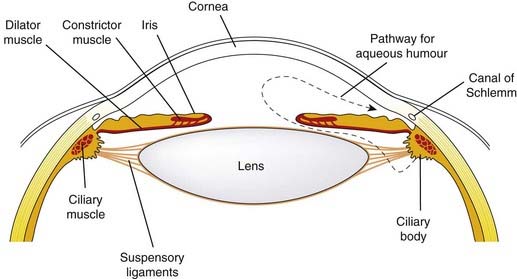
Fig. 13.5 The anterior chamber of the eye, showing the pathway for secretion and drainage of the aqueous humour.
In addition to these peripheral effects, muscarinic agonists that are able to penetrate the blood–brain barrier produce marked central effects due to activation mainly of M1 receptors in the brain. These include tremor, hypothermia and increased locomotor activity, as well as improved cognition (see Ch. 38). M1-selective agonists (e.g. xanomeline) are being investigated for possible use in treating dementia (see Eglen 2006; Ch. 39).
Clinical use
Currently there are few important uses for muscarinic agonists (though there are expectations that new, more selective agents may prove useful in various CNS disorders, particularly dementia). Current clinical uses are summarized in the clinical box (p. 160).
Clinical uses of muscarinic agonists and related drugs 
Muscarinic Antagonists
Muscarinic receptor antagonists (parasympatholytic drugs; Table 13.5) are competitive antagonists whose chemical structures usually contain ester and basic groups in the same relationship as ACh, but they have a bulky aromatic group in place of the acetyl group. The two naturally occurring compounds, atropine and hyoscine (also known as scopolamine), are alkaloids found in solanaceous plants. The deadly nightshade (Atropa belladonna) contains mainly atropine, whereas the thorn apple (Datura stramonium) contains mainly hyoscine. These are tertiary ammonium compounds that are sufficiently lipid-soluble to be readily absorbed from the gut or conjunctival sac and, importantly, to penetrate the blood–brain barrier. Quaternary ammonium compounds, which have peripheral actions very similar to those of atropine but, because of their exclusion from the brain, lack central actions, include hyoscine butylbromide and propantheline. Ipratropium, another quaternary ammonium compound, is used by inhalation as a bronchodilator. Cyclopentolate and tropicamide are tertiary amines developed for ophthalmic use and administered as eye drops. Pirenzepine is a relatively selective M1 receptor antagonist. Oxybutynin, tolterodine and darifenacin (M3-selective) are drugs that act on the bladder to inhibit micturition, and are used for treating urinary incontinence. They produce unwanted effects typical of muscarinic antagonists, such as dry mouth, constipation and blurred vision, but these are less severe than with earlier drugs.
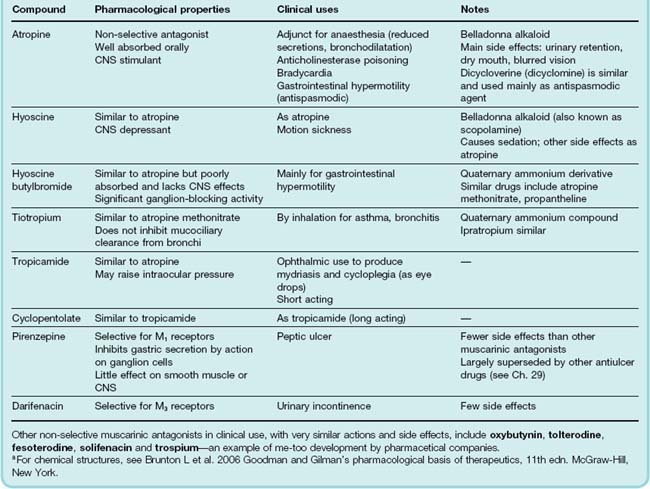
Effects of muscarinic antagonists
All the muscarinic antagonists produce basically similar peripheral effects, although some show a degree of selectivity, for example for the heart or bladder, reflecting heterogeneity among mAChRs (see p. 153).
The main effects of atropine are as follow.
Inhibition of secretions
Salivary, lacrimal, bronchial and sweat glands are inhibited by very low doses of atropine, producing an uncomfortably dry mouth and skin. Gastric secretion is only slightly reduced. Mucociliary clearance in the bronchi is inhibited, so that residual secretions tend to accumulate in the lungs. Ipratropium lacks this effect.
Effects on heart rate
Atropine causes tachycardia through block of cardiac mAChRs. The tachycardia is modest, up to 80–90 beats/min in humans. This is because there is no effect on the sympathetic system, but only inhibition of the existing parasympathetic tone. Tachycardia is most pronounced in young people, in whom vagal tone at rest is highest; it is often absent in the elderly. At very low doses, atropine causes a paradoxical bradycardia, possibly due to a central action. The response of the heart to exercise is unaffected. Arterial blood pressure is unaffected, because most resistance vessels have no cholinergic innervation.
Effects on the eye
The pupil is dilated (mydriasis) by atropine administration, and becomes unresponsive to light. Relaxation of the ciliary muscle causes paralysis of accommodation (cycloplegia), so that near vision is impaired. Intraocular pressure may rise; although this is unimportant in normal individuals, it can be dangerous in patients suffering from narrow-angle glaucoma.
Effects on the gastrointestinal tract
Gastrointestinal motility is inhibited by atropine, although this requires larger doses than the other effects listed, and is not complete. This is because excitatory transmitters other than ACh are important in normal function of the myenteric plexus (see Ch. 12). Atropine is used in pathological conditions in which there is increased gastrointestinal motility. Pirenzepine, owing to its selectivity for M1 receptors, inhibits gastric acid secretion in doses that do not affect other systems.
Effects on other smooth muscle
Bronchial, biliary and urinary tract smooth muscle are all relaxed by atropine. Reflex bronchoconstriction (e.g. during anaesthesia) is prevented by atropine, whereas bronchoconstriction caused by local mediators, such as histamine and leukotrienes (e.g. in asthma; Ch. 27) is unaffected. Biliary and urinary tract smooth muscle are only slightly affected in normal individuals, probably because transmitters other than ACh (see Ch. 12) are important in these organs; nevertheless, atropine and similar drugs commonly precipitate urinary retention in elderly men with prostatic enlargement. Incontinence due to bladder overactivity is reduced by muscarinic antagonists.
Effects on the CNS
Atropine produces mainly excitatory effects on the CNS. At low doses, this causes mild restlessness; higher doses cause agitation and disorientation. In atropine poisoning, which occurs mainly in young children who eat deadly nightshade berries, marked excitement and irritability result in hyperactivity and a considerable rise in body temperature, which is accentuated by the loss of sweating. These central effects are the result of blocking mAChRs in the brain, and they are opposed by anticholinesterase drugs such as physostigmine, which is an effective antidote to atropine poisoning. Hyoscine in low doses causes marked sedation, but has similar effects in high dosage. Hyoscine also has a useful antiemetic effect and is used in treating motion sickness. Muscarinic antagonists also affect the extrapyramidal system, reducing the involuntary movement and rigidity of patients with Parkinson’s disease (Ch. 39) and counteracting the extrapyramidal side effects of many antipsychotic drugs (Ch. 45).
Drugs acting on muscarinic receptors
Clinical use
The main uses of muscarinic antagonists (Table 13.5; clinical box, p. 162) are:
Apart from pirenzepine (M1-selective) and darifenacin (M3-selective), currently used muscarinic antagonists show little subtype selectivity, and differ mainly in their pharmacokinetic behaviour.
Clinical uses of muscarinic antagonists
Cardiovascular
Respiratory
Anaesthetic premedication
Gastrointestinal
Drugs Affecting Autonomic Ganglia
Ganglion Stimulants
Most nAChR agonists (apart from acetylcholine itself) act on either ganglionic and CNS receptors or at the motor endplate (Table 13.6).
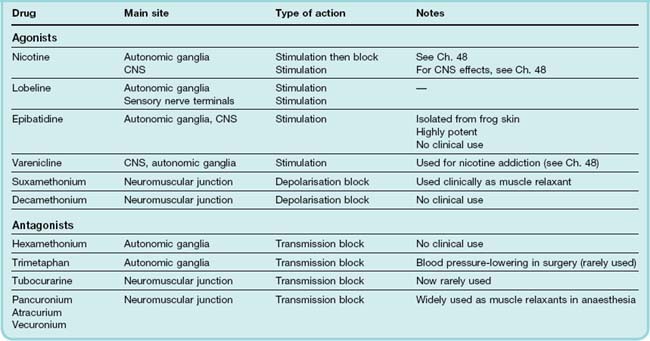
Nicotine and lobeline are tertiary amines found in the leaves of tobacco and lobelia plants, respectively. Nicotine belongs in pharmacological folklore, as it was the substance on the tip of Langley’s paintbrush causing stimulation of muscle fibres when applied to the endplate region, leading him to postulate in 1905 the existence of a ‘receptive substance’ on the surface of the fibres (Ch. 12). Epibatidine, found in the skin of poisonous frogs, is a highly potent nicotinic agonist selective for ganglionic and CNS receptors. It was found, unexpectedly, to be a powerful analgesic (see Ch. 41), though its autonomic side effects ruled out its clinical use. Varenicline, a synthetic agonist relatively selective for CNS receptors, is used (as is nicotine itself) to treat nicotine addiction (Ch. 48). Otherwise these drugs are used only as experimental tools.
They cause complex peripheral responses associated with generalised stimulation of autonomic ganglia. The effects of nicotine on the gastrointestinal tract and sweat glands are familiar to neophyte smokers (see Ch. 48), although usually insufficient to act as an effective deterrent.
Ganglion-Blocking Drugs
Ganglion block is often used in experimental studies on the autonomic nervous system but is of little clinical importance. It can occur by several mechanisms:
Sixty years ago, Paton and Zaimis investigated a series of linear bisquaternary compounds. Compounds with five or six carbon atoms (hexamethonium; Table 13.6) in the methylene chain linking the two quaternary groups produced ganglionic block, whereas compounds with nine or ten carbon atoms (decamethonium) produced neuromuscular block.5
Hexamethonium, although no longer used, deserves recognition as the first effective antihypertensive agent (see Ch. 22). The only ganglion-blocking drug currently in clinical use is trimetaphan (Table 13.6; see below).
Effects of ganglion-blocking drugs
The effects of ganglion-blocking drugs are numerous and complex, as would be expected, because both divisions of the autonomic nervous system are blocked indiscriminately. The description by Paton of ‘hexamethonium man’ cannot be bettered:
He is a pink-complexioned person, except when he has stood in a queue for a long time, when he may get pale and faint. His handshake is warm and dry. He is a placid and relaxed companion; for instance he may laugh but he can’t cry because the tears cannot come. Your rudest story will not make him blush, and the most unpleasant circumstances will fail to make him turn pale. His collars and socks stay very clean and sweet. He wears corsets and may, if you meet him out, be rather fidgety (corsets to compress his splanchnic vascular pool, fidgety to keep the venous return going from his legs). He dislikes speaking much unless helped with something to moisten his dry mouth and throat. He is long-sighted and easily blinded by bright light. The redness of his eyeballs may suggest irregular habits and in fact his head is rather weak. But he always behaves like a gentleman and never belches or hiccups. He tends to get cold and keeps well wrapped up. But his health is good; he does not have chilblains and those diseases of modern civilization, hypertension and peptic ulcer, pass him by. He gets thin because his appetite is modest; he never feels hunger pains and his stomach never rumbles. He gets rather constipated so that his intake of liquid paraffin is high. As old age comes on, he will suffer from retention of urine and impotence, but frequency, precipitancy and strangury will not worry him. One is uncertain how he will end, but perhaps if he is not careful, by eating less and less and getting colder and colder, he will sink into a symptomless, hypoglycaemic coma and die, as was proposed for the universe, a sort of entropy death.
(From Paton W D M 1954 The principles of ganglion block. Lectures on the scientific basis of medicine, vol. 2.)
In practice, the main effect is marked fall in arterial blood pressure resulting mainly from block of sympathetic ganglia, which causes arteriolar vasodilatation, and the block of cardiovascular reflexes. In particular, the venoconstriction, which occurs normally when a subject stands up and which is necessary to prevent the central venous pressure (and hence cardiac output) from falling sharply, is reduced. Standing thus causes a sudden fall in arterial pressure (postural hypotension) that can cause fainting. Similarly, the vasodilatation of skeletal muscle during exercise is normally accompanied by vasoconstriction elsewhere (e.g. splanchnic area) produced by sympathetic activity. If this adjustment is prevented, the overall peripheral resistance falls and the blood pressure also falls (postexercise hypotension).
Drugs acting on autonomic ganglia
Ganglion-stimulating drugs
Clinical use
Ganglion-blocking drugs, because of their many side effects, are obsolete as hypotensive agents, with the exception of trimetaphan, a very short-acting drug occasionally used as an intravenous infusion to produce controlled hypotension and minimise bleeding during certain types of surgery. Trimetaphan can also be used to lower blood pressure as an emergency procedure.
Neuromuscular-Blocking Drugs
The pharmacology of neuromuscular function is well reviewed by Bowman (1990). Drugs can block neuromuscular transmission either by acting presynaptically to inhibit ACh synthesis or release, or by acting postsynaptically, the latter being the site of action of all the clinically important drugs (except for botulinum toxin; see below).
Clinically, neuromuscular block is used only as an adjunct to anaesthesia, when artificial ventilation is available; it is not a therapeutic intervention. The drugs that are used all work postsynaptically, either (a) by blocking ACh receptors (or in some cases the ion channel) or (b) by activating ACh receptors and thus causing persistent depolarisation of the motor endplate. Apart from suxamethonium (see below) all of the drugs used clinically are non-depolarising agents.
Non-Depolarising Blocking Agents
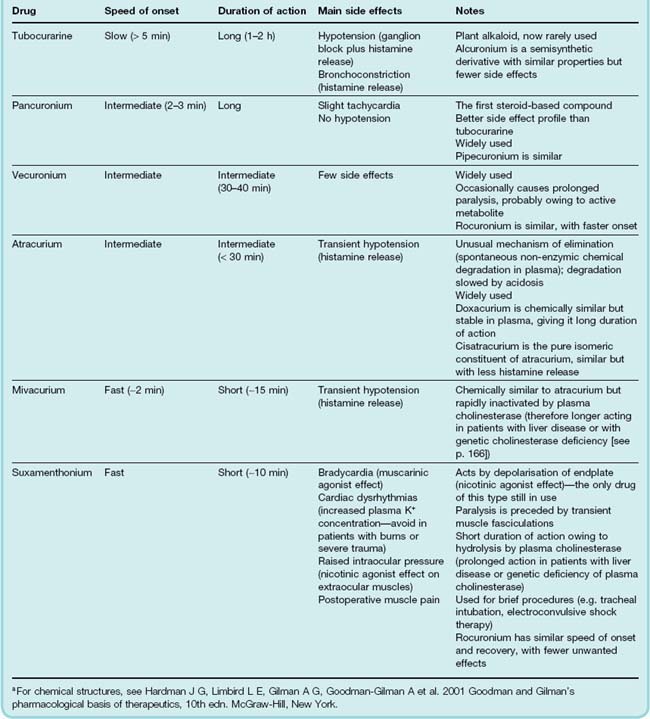
In 1856, Claude Bernard, in a famous experiment, showed that ‘curare’ causes paralysis by blocking neuromuscular transmission, rather than by abolishing nerve conduction or muscle contractility. Curare is a mixture of naturally occurring alkaloids found in various South American plants and used as arrow poisons by South American Indians. The most important component is tubocurarine, which is now rarely used in clinical medicine, being superseded by synthetic drugs with improved properties. The most important are pancuronium, vecuronium and atracurium (Table 13.7), which differ mainly in their duration of action. Gallamine was the first useful synthetic successor to tubocurarine, but has been replaced by compounds with fewer side effects. These substances are all quaternary ammonium compounds, which means that they are poorly absorbed and generally rapidly excreted. They also fail to cross the placenta, which is important in relation to their use in obstetric anaesthesia. The low oral absorption of tubocurarine allowed it to be used safely in the hunting of animals for food, leaving the meat safe to eat.
Mechanism of action
Non-depolarising blocking agents all act as competitive antagonists (see Ch. 2) at the ACh receptors of the endplate.
The amount of ACh released by a nerve impulse normally exceeds by several-fold what is needed to elicit an action potential in the muscle fibre. It is therefore necessary to block 70–80% of the receptor sites before transmission actually fails. In any individual muscle fibre, transmission is all or nothing, so graded degrees of block represent a varying proportion of muscle fibres failing to respond. In this situation, where the amplitude of epp in all the fibres is close to threshold (just above in some, just below in others), small variations in the amount of transmitter released, or in the rate at which it is destroyed, will have a large effect on the proportion of fibres contracting, so the degree of block is liable to vary according to various physiological circumstances (e.g. stimulation frequency, temperature and cholinesterase inhibition), which normally have relatively little effect on the efficiency of transmission.
Non-depolarising blocking agents also block facilitatory presynaptic autoreceptors, and thus inhibit the release of ACh during repetitive stimulation of the motor nerve (see Prior et al., 1995), resulting in the phenomenon of ‘tetanic fade’, which is often used by anaesthetists to monitor postoperative recovery of neuromuscular transmission (see p. 166).
Effects of non-depolarising blocking drugs
The effects of non-depolarising neuromuscular-blocking agents are mainly due to motor paralysis, although some of the drugs also produce clinically significant autonomic effects.
The first muscles to be affected are the extrinsic eye muscles (causing double vision) and the small muscles of the face, limbs and pharynx (causing difficulty in swallowing). Respiratory muscles are the last to be affected and the first to recover. An experiment in 1947 in which a heroic volunteer was fully curarised under artificial ventilation established this orderly paralytic march, and showed that consciousness and awareness of pain were quite normal even when paralysis was complete.6
Unwanted effects
The main side effect of tubocurarine is a fall in arterial pressure, due to (a) ganglion block and (b) histamine release from mast cells (see Ch. 17), which can also give rise to bronchospasm in sensitive individuals. This is unrelated to nAChRs but also occurs with atracurium and mivacurium (as well as with some unrelated drugs such as morphine; see Ch. 41). The other non-depolarising blocking drugs lack these side effects, and hence cause less hypotension. Pancuronium (and also gallamine, now obsolete) blocks mAChRs, particularly in the heart, which results in tachycardia.
Pharmacokinetic aspects
Neuromuscular-blocking agents are used mainly in anaesthesia to produce muscle relaxation. They are given intravenously but differ in their rates of onset and recovery (Fig. 13.6 and Table 13.7).
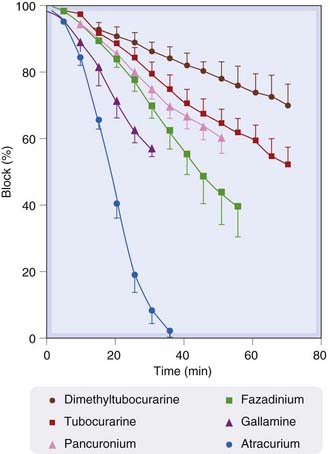
Fig. 13.6 Rate of recovery from various non-depolarising neuromuscular-blocking drugs in humans.
Drugs were given intravenously to patients undergoing surgery, in doses just sufficient to cause 100% block of the tetanic tension of the indirectly stimulated adductor pollicis muscle. Recovery of tension was then followed as a function of time.
(From Payne J P, Hughes R 1981 Br J Anaesth 53: 45.)
Most of the non-depolarising blocking agents are metabolised by the liver or excreted unchanged in the urine, exceptions being atracurium, which hydrolyses spontaneously in plasma, and mivacurium, which, like suxamethonium (see below), is hydrolysed by plasma cholinesterase. Their duration of action varies between about 15 min and 1–2 h (Table 13.7), by which time the patient regains enough strength to cough and breathe properly. The route of elimination is important, because many patients undergoing anaesthesia have impaired renal or hepatic function, which, depending on the drug used, can enhance or prolong the paralysis to an important degree.
Atracurium was designed to be chemically unstable at physiological pH (splitting into two inactive fragments by cleavage at one of the quaternary nitrogen atoms), although indefinitely stable when stored at an acid pH. It has a short duration of action, which is unaffected by renal or hepatic function. Because of the marked pH dependence of its degradation, however, its action becomes considerably briefer during respiratory alkalosis caused by hyperventilation.
Rapid postoperative recovery of muscle strength is needed to reduce the risk of complications. The cholinesterase inhibitor, neostigmine (see p. 169) is often used to reverse the action of non-depolarising drugs postoperatively. Co-administration of atropine is necessary to prevent unwanted parasympathomimetic effects. An alternative approach currently in development (see Nicholson et al., 2007) is the use of a synthetic cyclodextrin, sugammadex, a macromolecule that selectively binds steroidal neuromuscular blocking drugs such as pancuronium as an inactive complex in the plasma. The complex is excreted unchanged in the urine. Sugammadex is claimed to produce more rapid reversal of block with fewer unwanted effects than neostigmine.
Depolarising Blocking Agents
This class of neuromuscular-blocking drugs was discovered by Paton and Zaimis in their study of the effects of symmetrical bisquaternary ammonium compounds. One of these, decamethonium, was found to cause paralysis without appreciable ganglion-blocking activity. Several features of its action showed it to be different from competitive blocking drugs such as tubocurarine. In particular, it was found to produce a transient twitching of skeletal muscle (fasciculation) before causing block, and when it was injected into chicks it caused a powerful extensor spasm,7 whereas tubocurarine simply caused flaccid paralysis. In 1951, Burns and Paton showed that it acted as an agonist, causing a maintained depolarisation at the endplate region of the muscle fibre, which led to a loss of electrical excitability (see p. 156), and they coined the term depolarisation block. Fasciculation occurs because the developing endplate depolarisation initially causes a discharge of action potentials in the muscle fibre. This subsides after a few seconds as the electrical excitability of the endplate region of the fibre is lost. Decamethonium itself was used clinically but has the disadvantage of too long a duration of action.
Suxamethonium (Table 13.7)—the only depolarising blocking drug currently used—is closely related in structure to both decamethonium and ACh (consisting of two ACh molecules linked by their acetyl groups) and acts similarly, but its action is brief, because it is quickly hydrolysed by plasma cholinesterase. When given intravenously, however, its depolarising action lasts for long enough to cause the endplate region of the muscle fibres to become inexcitable. ACh, in contrast, when released from the nerve, reaches the endplate in very brief spurts and is rapidly hydrolysed in situ, so it never causes sufficiently prolonged depolarisation to result in block. If cholinesterase is inhibited, however (see p. 171), it is possible for the circulating ACh concentration to reach a level sufficient to cause depolarisation block.
Comparison of non-depolarising and depolarising blocking drugs
There are several differences in the pattern of neuromuscular block produced by depolarising and non-depolarising mechanisms:
Unwanted effects and dangers of suxamethonium
Suxamethonium has several unwanted, and in some cases dangerous, side effects (see Table 13.7), but remains in use because of the rapid recovery that follows its withdrawal—significantly more rapid than the recovery from non-depolarising agents.
Potassium release
The increase in cation permeability of the motor endplates causes a net loss of K+ from muscle, and thus a small rise in plasma K+ concentration. In normal individuals, this is not important, but in cases of trauma, especially burns or injuries causing muscle denervation, it may be (Fig. 13.7). This is because denervation causes ACh receptors to spread to regions of the muscle fibre away from the endplates (see Ch. 12), so that a much larger area of membrane is sensitive to suxamethonium. The resulting hyperkalaemia can be enough to cause ventricular dysrhythmia or even cardiac arrest.
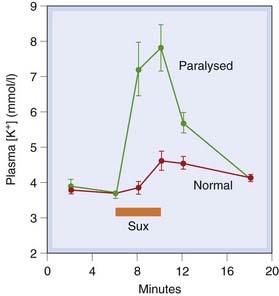
Fig. 13.7 Effect of suxamethonium (Sux) on plasma potassium concentration in humans.
Blood was collected from veins draining paralysed and non-paralysed limbs of seven injured patients undergoing surgery. The injuries had resulted in motor nerve degeneration, and hence denervation supersensitivity of the affected muscles.
(From Tobey R E et al. 1972 Anaesthesiology 37: 322.)
Increased intraocular pressure
This results from contracture of extraocular muscles applying pressure to the eyeball. It is particularly important to avoid this if the eyeball has been injured.
Prolonged paralysis
The action of suxamethonium given intravenously normally lasts for less than 5 min, because the drug is hydrolysed by plasma cholinesterase. Its action is prolonged by various factors that reduce the activity of this enzyme:
Malignant hyperthermia
This is a rare inherited condition, due to a mutation of the Ca2+ release channel of the sarcoplasmic reticulum (the ryanodine receptor, see Ch. 4), which results in intense muscle spasm and a dramatic rise in body temperature when certain drugs are given (see Ch. 11). The most commonly implicated drugs are suxamethonium and halothane (see Ch. 40), although it can be precipitated by a variety of other drugs. The condition carries a very high mortality (about 65%) and is treated by administration of dantrolene, a drug that inhibits muscle contraction by preventing Ca2+ release from the sarcoplasmic reticulum.
Neuromuscular-blocking drugs
Drugs That Act Presynaptically
Drugs That Inhibit Acetylcholine Synthesis
The steps in the synthesis of ACh in the presynaptic nerve terminals are shown in Figure 13.2. The rate-limiting process appears to be the transport of choline into the nerve terminal. Hemicholinium blocks this transport and thereby inhibits ACh synthesis. It is useful as an experimental tool but has no clinical applications. Its blocking effect on transmission develops slowly, as the existing stores of ACh become depleted. Vesamicol, which acts by blocking ACh transport into synaptic vesicles, has a similar effect.
Drugs That Inhibit Acetylcholine Release
Acetylcholine release by a nerve impulse involves the entry of Ca2+ into the nerve terminal; the increase in [Ca2+]i stimulates exocytosis and increases the rate of quantal release (Fig. 13.2). Agents that inhibit Ca2+ entry include Mg2+ and various aminoglycoside antibiotics (e.g. streptomycin and neomycin; see Ch. 50), which occasionally produce muscle paralysis as an unwanted side effect when used clinically.
Two potent neurotoxins, namely botulinum toxin and β-bungarotoxin, act specifically to inhibit ACh release. Botulinum toxin is a protein produced by the anaerobic bacillus Clostridium botulinum, an organism that can multiply in preserved food and can cause botulism, an extremely serious type of food poisoning.8
The potency of botulinum toxin is extraordinary, the minimum lethal dose in a mouse being less than 10−12 g—only a few million molecules. It belongs to the group of potent bacterial exotoxins that includes tetanus and diphtheria toxins. They possess two subunits, one of which binds to a membrane receptor and is responsible for cellular specificity. By this means, the toxin enters the cell, where the other subunit produces the toxic effect (see Montecucco & Schiavo, 1995). Botulinum toxin contains several components (A–G). They are peptidases that cleave specific proteins involved in exocytosis (synaptobrevins, syntaxins, etc.; see Ch. 4), thereby producing a long-lasting block of synaptic function. Each toxin component inactivates a different functional protein—a remarkably coordinated attack by a humble bacterium on a vital component of mammalian physiology.
Botulinum poisoning causes progressive parasympathetic and motor paralysis, with dry mouth, blurred vision and difficulty in swallowing, followed by progressive respiratory paralysis. Treatment with antitoxin is effective only if given before symptoms appear, for once the toxin is bound its action cannot be reversed. Mortality is high, and recovery takes several weeks. Anticholinesterases and drugs that increase transmitter release (see p. 172) are ineffective in restoring transmission. Botulinum toxin, given by local injection, has a number of clinical uses, including:
Injections need to be repeated every few months. Botulinum toxin is antigenic, and may lose its effectiveness due to its immunogenicity. There is a risk of more general muscle paralysis if the toxin spreads beyond the injected region.
Botox, as well as being a potential biological weapon of war, is famously fashionable as a wrinkle remover—reflecting strangely on the modern world. It removes frown lines by paralysing the superficial muscles that pucker the skin.
β-Bungarotoxin is a protein contained in the venom of various snakes of the cobra family, and has a similar action to botulinum toxin, although its active component is a phospholipase rather than a peptidase. The same venoms also contain α-bungarotoxin (see p. 23), which blocks postsynaptic ACh receptors. These snakes evidently cover all eventualities as far as causing paralysis of their victims is concerned.
Drugs That Enhance Cholinergic Transmission
Drugs that enhance cholinergic transmission act either by inhibiting cholinesterase (the main group) or by increasing ACh release. In this chapter, we focus on the peripheral actions of such drugs; drugs affecting cholinergic transmission in the CNS, used to treat senile dementia, are discussed in Chapter 39.
Distribution and Function of Cholinesterase
There are two distinct types of cholinesterase, namely acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE), closely related in molecular structure but differing in their distribution, substrate specificity and functions (see Chatonnet & Lockridge, 1989). Both consist of globular catalytic subunits, which constitute the soluble forms found in plasma (BuChE) and cerebrospinal fluid (AChE). Elsewhere, the catalytic units are linked to accessory proteins, which tether them like a bunch of balloons to the basement membrane (at the neuromuscular junction) or to the neuronal membrane at neuronal synapses (and also, oddly, the erythrocyte membrane, where the function of the enzyme is unknown).
The bound AChE at cholinergic synapses serves to hydrolyse the released transmitter and terminate its action rapidly. Soluble AChE is also present in cholinergic nerve terminals, where it has a role in regulating the free ACh concentration, and from which it may be secreted; the function of the secreted enzyme is so far unclear. AChE is quite specific for ACh and closely related esters such as methacholine. Certain neuropeptides, such as substance P (Ch. 19) are inactivated by AChE, but it is not known whether this is of physiological significance. Overall, there is poor correspondence between the distribution of cholinergic synapses and that of AChE, both in the brain and in the periphery, and AChE most probably has synaptic functions other than disposal of ACh, although the details remain unclear (see review by Silman & Sussman, 2005; Zimmerman & Soreq, 2006).
Butyrylcholinesterase (BuChE, or pseudocholinesterase) has a widespread distribution, being found in tissues such as liver, skin, brain and gastrointestinal smooth muscle, as well as in soluble form in the plasma. It is not particularly associated with cholinergic synapses, and its physiological function is unclear. It has a broader substrate specificity than AChE. It hydrolyses the synthetic substrate butyrylcholine more rapidly than ACh, as well as other esters, such as procaine, suxamethonium and propanidid (a short-acting anaesthetic agent; see Ch. 40). The plasma enzyme is important in relation to the inactivation of the drugs listed above. Genetic variants of BuChE causing significantly reduced enzymic activity occur rarely (see above and Ch. 11), and these partly account for the variability in the duration of action of these drugs. The very short duration of action of ACh given intravenously (see Fig. 13.1) results from its rapid hydrolysis in the plasma. Normally, AChE and BuChE between them keep the plasma ACh at an undetectably low level, so ACh (unlike noradrenaline) is strictly a neurotransmitter and not a hormone.
Both AChE and BuChE belong to the class of serine hydrolases, which includes many proteases such as trypsin. The active site of AChE comprises two distinct regions (Fig. 13.8): an anionic site (glutamate residue), which binds the basic (choline) moiety of ACh; and an esteratic (catalytic) site (histidine + serine). As with other serine hydrolases, the acidic (acetyl) group of the substrate is transferred to the serine hydroxyl group, leaving (transiently) an acetylated enzyme molecule and a molecule of free choline. Spontaneous hydrolysis of the serine acetyl group occurs rapidly, and the overall turnover number of AChE is extremely high (over 10 000 molecules of ACh hydrolysed per second by a single active site).
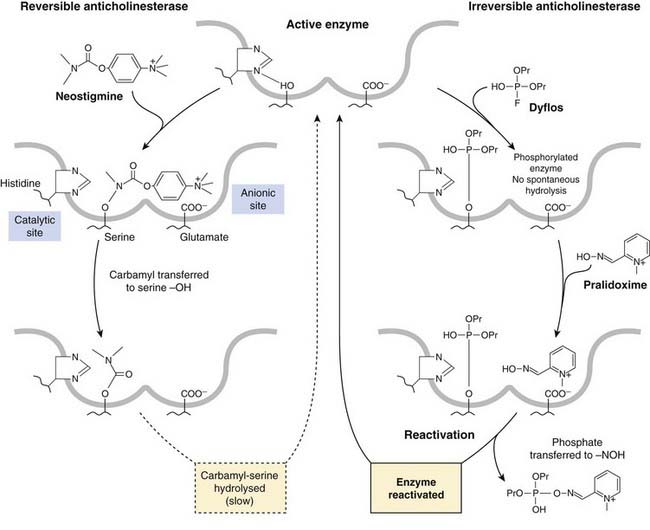
Fig. 13.8 Action of anticholinesterase drugs.
Reversible anticholinesterase (neostigmine): recovery of activity by hydrolysis of the carbamylated enzyme takes many minutes. Irreversible anticholinesterase (dyflos): reactivation of phosphorylated enzyme by pralidoxime. The representation of the active site is purely diagrammatic and by no means representative of the actual molecular structure.
Drugs That Inhibit Cholinesterase
Peripherally acting anticholinesterase drugs, summarized in Table 13.8, fall into three main groups according to the nature of their interaction with the active site, which determines their duration of action. Most of them inhibit AChE and BuChE about equally. Centrally acting anticholinesterases, developed for the treatment of dementia, are discussed in Chapter 39.
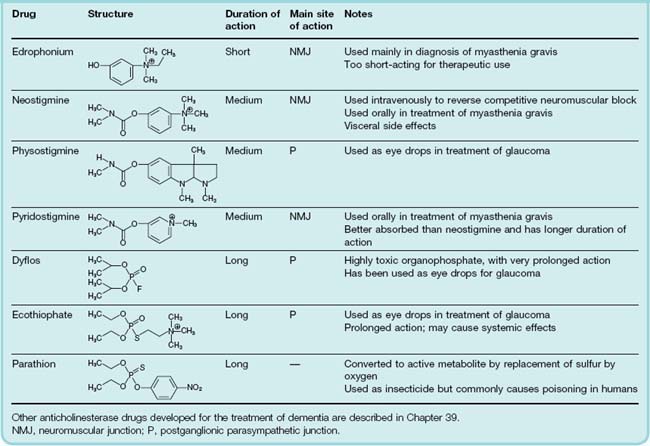
Short-acting anticholinesterases
The only important drug of this type is edrophonium, a quaternary ammonium compound that binds to the anionic site of the enzyme only. The ionic bond formed is readily reversible, and the action of the drug is very brief. It is used mainly for diagnostic purposes, because improvement of muscle strength by an anticholinesterase is characteristic of myasthenia gravis (see p. 171) but does not occur when muscle weakness is due to other causes.
Medium-duration anticholinesterases
These include neostigmine and pyridostigmine, which are quaternary ammonium compounds of clinical importance, and physostigmine (eserine), a tertiary amine, which occurs naturally in the Calabar bean.9
These drugs are all carbamyl, as opposed to acetyl, esters, and all possess basic groups that bind to the anionic site. Transfer of the carbamyl group to the serine hydroxyl group of the esteratic site occurs as with ACh, but the carbamylated enzyme is very much slower to hydrolyse (Fig. 13.8), taking minutes rather than microseconds. The anticholinesterase drug is therefore hydrolysed, but at a negligible rate compared with ACh, and the slow recovery of the carbamylated enzyme means that the action of these drugs is quite long-lasting.
Irreversible anticholinesterases
Irreversible anticholinesterases (Table 13.8) are pentavalent phosphorus compounds containing a labile group such as fluoride (in dyflos) or an organic group (in parathion and ecothiophate). This group is released, leaving the serine hydroxyl group of the enzyme phosphorylated (Fig. 13.8). Most of these organophosphate compounds, of which there are many, were developed as war gases and pesticides as well as for clinical use; they interact only with the esteratic site of the enzyme and have no cationic group. Ecothiophate is an exception in having a quaternary nitrogen group designed to bind also to the anionic site.
The inactive phosphorylated enzyme is usually very stable. With drugs such as dyflos, no appreciable hydrolysis occurs, and recovery of enzymic activity depends on the synthesis of new enzyme molecules, a process that may take weeks. With other drugs, such as ecothiophate, slow hydrolysis occurs over the course of a few days, so that their action is not strictly irreversible. Dyflos and parathion are volatile non-polar substances of very high lipid solubility, and are rapidly absorbed through mucous membranes and even through unbroken skin and insect cuticles; the use of these agents as war gases or insecticides relies on this property. The lack of a specificity-conferring quaternary group means that most of these drugs block other serine hydrolases (e.g. trypsin, thrombin), although their pharmacological effects result mainly from cholinesterase inhibition.
Effects of anticholinesterase drugs
Cholinesterase inhibitors affect peripheral as well as central cholinergic synapses.
Some organophosphate compounds can produce, in addition, a severe form of neurotoxicity.
Effects on autonomic cholinergic synapses
These mainly reflect enhancement of ACh activity at parasympathetic postganglionic synapses (i.e. increased secretions from salivary, lacrimal, bronchial and gastrointestinal glands; increased peristaltic activity; bronchoconstriction; bradycardia and hypotension; pupillary constriction; fixation of accommodation for near vision; fall in intraocular pressure). Large doses can stimulate, and later block, autonomic ganglia, producing complex autonomic effects. The block, if it occurs, is a depolarisation block and is associated with a build-up of ACh in the plasma and body fluids. Neostigmine and pyridostigmine tend to affect neuromuscular transmission more than the autonomic system, whereas physostigmine and organophosphates show the reverse pattern. The reason is not clear, but therapeutic usage takes advantage of this partial selectivity.
Acute anticholinesterase poisoning (e.g. from contact with insecticides or war gases) causes severe bradycardia, hypotension and difficulty in breathing. Combined with a depolarising neuromuscular block and central effects (see below), the result may be fatal.
Effects on the neuromuscular junction
The twitch tension of a muscle stimulated via its motor nerve is increased by anticholinesterases, owing to repetitive firing in the muscle fibre associated with prolongation of the epp. Normally, the ACh is hydrolysed so quickly that each stimulus initiates only one action potential in the muscle fibre, but when AChE is inhibited this is converted to a short train of action potentials in the muscle fibre, and hence greater tension. Much more important is the effect produced when transmission has been blocked by a non-depolarising blocking agent such as pancuronium. In this case, addition of an anticholinesterase can dramatically restore transmission. If a large proportion of the receptors is blocked, the majority of ACh molecules will normally encounter, and be destroyed by, an AChE molecule before reaching a vacant receptor; inhibiting AChE gives the ACh molecules a greater chance of finding a vacant receptor before being destroyed, and thus increase the epp so that it reaches threshold. In myasthenia gravis (see below), transmission fails because there are too few ACh receptors, and cholinesterase inhibition improves transmission just as it does in curarised muscle.
In large doses, such as can occur in poisoning, anticholinesterases initially cause twitching of muscles. This is because spontaneous ACh release can give rise to epps that reach the firing threshold. Later, paralysis may occur due to depolarisation block, which is associated with the build-up of ACh in the plasma and tissue fluids.
Effects on the CNS
Tertiary compounds, such as physostigmine, and the non-polar organophosphates penetrate the blood–brain barrier freely and affect the brain. The result is an initial excitation, which can result in convulsions, followed by depression, which can cause unconsciousness and respiratory failure. These central effects result mainly from the activation of mAChRs, and are antagonised by atropine. The use of anticholinesterases to treat senile dementia is discussed in Chapter 39.
Cholinesterase and anticholinesterase drugs
Neurotoxicity of organophosphates
Many organophosphates can cause a severe, though rare, type of delayed peripheral nerve degeneration, leading to progressive weakness and sensory loss. This is not a problem with clinically used anticholinesterases but occasionally results from poisoning with insecticides or nerve gases. In 1931, an estimated 20 000 Americans were affected, some fatally, by contamination of fruit juice with an organophosphate insecticide, and other similar outbreaks have been recorded. The mechanism of this reaction is only partly understood, but it seems to result from inhibition of a neuropathy target esterase distinct from cholinesterase.
The main uses of anticholinesterases are summarised in the clinical box, above.
Clinical uses of anticholinesterase drugs
Cholinesterase Reactivation
Spontaneous hydrolysis of phosphorylated cholinesterase is extremely slow, a fact that makes poisoning with organophosphates very dangerous. Pralidoxime (Fig. 13.8) reactivates the enzyme by bringing an oxime group into close proximity with the phosphorylated esteratic site. This group is a strong nucleophile and lures the phosphate group away from the serine hydroxyl group of the enzyme. The effectiveness of pralidoxime in reactivating plasma cholinesterase activity in a poisoned subject is shown in Figure 13.9. The main limitation to its use as an antidote to organophosphate poisoning is that, within a few hours, the phosphorylated enzyme undergoes a chemical change (‘ageing’) that renders it no longer susceptible to reactivation, so that pralidoxime must be given early in order to work. Pralidoxime does not enter the brain, but related compounds have been developed to treat the central effects of organophosphate poisoning.
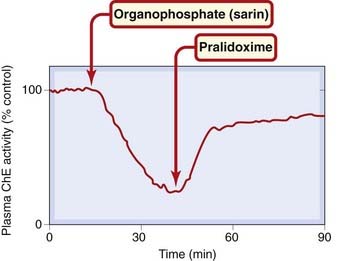
Fig. 13.9 Reactivation of plasma cholinesterase (ChE) in a volunteer subject by intravenous injection of pralidoxime.
(Redrawn from Sim V M 1965 JAMA 192: 404.)
Myasthenia gravis
The neuromuscular junction is a robust structure that very rarely fails, myasthenia gravis being one of the very few disorders that specifically affects it (see Lindstrom, 2000). This disease affects about 1 in 2000 individuals, who show muscle weakness and increased fatiguability resulting from a failure of neuromuscular transmission. The tendency for transmission to fail during repetitive activity can be seen in Figure 13.10. Functionally, it results in the inability of muscles to produce sustained contractions, of which the characteristic drooping eyelids of myasthenic patients are a sign. The effectiveness of anticholinesterase drugs in improving muscle strength in myasthenia was discovered in 1931, long before the cause of the disease was known.
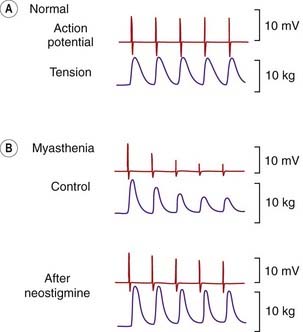
Fig. 13.10 Neuromuscular transmission in a normal and a myasthenic human subject.
Electrical activity was recorded with a needle electrode in the adductor pollicis muscle, in response to ulnar nerve stimulation (3 Hz) at the wrist. In a normal subject, electrical and mechanical response is well sustained. In a myasthenic patient, transmission fails rapidly when the nerve is stimulated. Treatment with neostigmine improves transmission.
(From Desmedt J E 1962 Bull Acad Roy Med Belg VII 2: 213.)
The cause of the transmission failure is an autoimmune response that causes a loss of nAChRs from the neuromuscular junction, first revealed in studies showing that the number of bungarotoxin-binding sites at the endplates of myasthenic patients was reduced by about 70% compared with normal. It had been suspected that myasthenia had an immunological basis, because removal of the thymus gland was frequently of benefit. Immunisation of rabbits with purified ACh receptor causes, after a delay, symptoms very similar to those of human myasthenia gravis. The presence of antibody directed against the ACh receptor protein can be detected in the serum of myasthenic patients, but the reason for the development of the autoimmune response in humans is still unknown (see Lindstrom, 2000).
The improvement of neuromuscular function by anticholinesterase treatment (shown in Fig. 13.10) can be dramatic, but if the disease progresses too far, the number of receptors remaining may become too few to produce an adequate epp, and anticholinesterase drugs will then cease to be effective.
Alternative approaches to the treatment of myasthenia are to remove circulating antibody by plasma exchange, which is transiently effective, or, for a more prolonged effect, to inhibit antibody production with steroids (e.g. prednisolone) or immunosuppressant drugs (e.g. azathioprine; see Ch. 26).
Other Drugs That Enhance Cholinergic Transmission
It was observed many years ago that tetraethylammonium, better known as a ganglion-blocking drug, could reverse the neuromuscular-blocking action of tubocurarine, and this was shown to be because it increases the release of transmitter evoked by nerve stimulation. Subsequently, aminopyridines, which block potassium channels (see Ch. 4), and thus prolong the action potential in the presynaptic nerve terminal, were found to act similarly and to be considerably more potent and selective in their actions than tetraethylammonium. These drugs are not selective for cholinergic nerves but increase the evoked release of many different transmitters, so have too many unwanted effects to be useful in treating neuromuscular disorders.
References and Further Reading
Nicholls J.G., Martin A.R., Wallace B.G., Fuchs P. From neuron to brain. Sunderland: Sinauer; 2001. (Excellent general textbook)
Caulfield M.P., Birdsall N.J. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol. Rev.. 1998;50:279-290. (The accepted definitions and characteristics of mAChRs)
Dajas-Bailador F., Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol. Sci.. 2004;25:217-324. (Short review article focusing on the presynaptic actions of nAChRs in the CNS and periphery)
Eglen R.M. Muscarinic receptor subtypes in neuronal and non-neuronal cholinergic function. Auton. Autacoid. Pharmacol.. 2006;26:219-233. (Covers the conventional roles of mAChRs in the autonomic system as well as possible more general involvement in immunological and other mechanisms)
Eglen R.M., Choppin A., Dillon M.P., Hedge S. Muscarinic receptor ligands and their therapeutic potential. Curr. Opin. Chem. Biol.. 1999;3:426-432. (Review of the future development of muscarinic agonists and antagonists for different indications)
Goyal R.K. Muscarinic receptor subtypes: physiology and clinical implications. New Engl. J. Med.. 1989;321:1022-1029. (Good general review)
Hogg R.C., Raggenbass M., Bertrand D. Nicotinic acetylcholine receptors: from structure to brain function. Rev. Physiol. Biochem. Pharmacol.. 2003;147:1-46. (Comprehensive review covering all aspects of nAChR structure and function)
Kalamida D., Poulas K., Avramopoulou V., et al. Muscle and neuronal nicotinic acetylcholine receptors: structure, function and pathogenicity. FEBS J.. 2007;274:3799-3845. (Excellent comprehensive review)
Wess J. Molecular biology of muscarinic acetylcholine receptors. Crit. Rev. Neurobiol.. 1996;10:69-99. (Describes receptor subtypes in detail)
Wess J., Eglen R.M., Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat. Rev. Drug Discov.. 2007;6:721-733. (Describes the subtle functional deficits in mice lacking particular receptor subtypes)
Wessler I., Kirkpatrick C.J. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br. J. Pharmacol.. 2008;154:1558-1571. (Summarises recent findings that reveal surprisingly diverse roles for acetylcholine)
Lindstrom J.M. Acetylcholine receptors and myasthenia. Muscle Nerve. 2000;23:453-477. (Good review article on nAChR subtypes and current views on the pathophysiology of myasthenia gravis and related neuromuscular disorders)
Parsons S.M., Prior C., Marshall I.G. Acetylcholine transport, storage and release. Int. Rev. Neurobiol.. 1993;35:279-390. (Useful review of the local metabolism of ACh)
Usdin T.B., Eiden L.E., Bonner T.I., Erickson J.D. Molecular biology of the vesicular ACh transporter. Trends Neurosci.. 1995;18:218-224. (Short review article)
Drugs affecting the neuromuscular junction
Bowman W.C. Pharmacology of neuromuscular function. Bristol: Wright; 1990. (Detailed textbook)
Montecucco C., Schiavo G. Structure and function of botulinum neurotoxins. Q. Rev. Biophys.. 1995;28:423-472. (Discusses the mode of action of an important group of presynaptic neurotoxins)
Nicholson W.T., Sprung J., Jankowski C.J. Sugammadex: a novel agent for the reversal of neuromuscular blockade. Pharmacotherapy. 2007;27:1181-1188. (An alternative to neostigmine)
Prior C., Tian L., Dempster J., Marshall I.G. Prejunctional actions of muscle relaxants: synaptic vesicles and transmitter mobilization as sites of action. Gen. Pharmacol.. 1995;26:659-666. (Emphasises the role of presynaptic inhibition in the action of neuromuscular-blocking drugs)
Chatonnet A., Lockridge O. Comparison of butyrylcholinesterase and acetylcholinesterase. Biochem. J.. 1989;260:625-634. (Short review on the nature and functions of cholinesterases)
Silman I., Sussman J.L. Acetylcholinesterase: ‘classical’ and ‘non-classical’ functions and pharmacology. Curr. Opin. Pharmacol.. 2005;5:293-302. (Review, covering molecular structure, as well as functions of cholinesterase)
Zimmerman G., Soreq H. Termination and beyond: acetylcholinesterase as a modulator of synaptic transmission. Cell Tissue Res.. 2006;326:655-669. (Speculative review of evidence suggesting functions for AChE other than ACh hydrolysis)
1Cevimeline was recently introduced to improve salivary and lacrimal secretion in Sjögren’s disease, a rare autoimmune disease causing loss of parasympathetic effects.
2Unlike most other antagonists, gallamine acts allosterically (i.e. at a site distinct from the ACh binding site).
3Acetylcholine is synthesised, stored and released by many non-neuronal cells, such as epithelia and cells of the immune system. The regulatory and trophic functions of non-neuronal ACh are a current topic of investigation (see Wessler & Kirkpatrick, 2008).
4Known in the USA as succinylcholine.
5Based on their structural similarity to ACh, these compounds were originally believed to act as competitive antagonists. However, they are now known to act mainly by blocking the ion channel rather than the receptor site itself.
6The risk of patients waking up paralysed during surgery is a serious worry for anaesthetists.
7Birds (and frogs) possess a special type of skeletal muscle, rare in mammals, that has many endplates scattered over the surface of each muscle fibre. Agonists cause a diffuse depolarisation in such muscles, resulting in a maintained contracture. In normal skeletal muscle, with only one endplate per fibre, endplate depolarisation is too localised to cause contracture on its own.
8Among the more spectacular outbreaks of botulinum poisoning was an incident on Loch Maree in Scotland in 1922, when all eight members of a fishing party died after eating duck pâté for their lunch. Their ghillies, consuming humbler fare no doubt, survived. The innkeeper committed suicide.
9Otherwise known as the ordeal bean. In the Middle Ages, extracts of these beans were used to determine the guilt or innocence of those accused of crime or heresy. Death implied guilt.