37 Amino acid transmitters
Overview
In this chapter, we discuss the major neurotransmitters in the central nervous system (CNS), namely the excitatory transmitter, glutamate, and the inhibitory transmitters, GABA and glycine. It is an area in which scientific interest has been intense in recent years. Unravelling the complexities of amino acid receptors and signalling mechanisms has thrown considerable light on their role in brain function and their likely involvement in CNS disease. Drugs that target specific receptors and transporters have been developed, but translating this knowledge into drugs for therapeutic use is only now beginning to happen. Here, we present the pharmacological principles and include recent references for those seeking more detail.
Excitatory Amino Acids
Excitatory Amino Acids as CNS Transmitters
L-Glutamate is the principal and ubiquitous excitatory transmitter in the CNS (see Cotman et al., 1995, for general review). Aspartate plays a similar role in certain brain regions, and possibly also homocysteate, but this is controversial.
 The realisation of glutamate’s importance came slowly (see Watkins & Jane, 2006). By the 1950s, work on the peripheral nervous system had highlighted the transmitter roles of acetylcholine and catecholamines, and as the brain also contained these substances, there seemed little reason to look further. The presence of γ-aminobutyric acid (GABA; see below) in the brain, and its powerful inhibitory effect on neurons, were discovered in the 1950s, and its transmitter role was postulated. At the same time, work by Curtis’s group in Canberra showed that glutamate and various other acidic amino acids produced a strong excitatory effect, but it seemed inconceivable that such workaday metabolites could actually be transmitters. Through the 1960s, GABA and excitatory amino acids (EAAs) were thought, even by their discoverers, to be mere pharmacological curiosities. In the 1970s, the humblest amino acid, glycine, was established as an inhibitory transmitter in the spinal cord, giving the lie to the idea that transmitters had to be exotic molecules, too beautiful for any role but to sink into the arms of a receptor. Once glycine had been accepted, the rest quickly followed. A major advance was the discovery of EAA antagonists, based on the work of Watkins in Bristol, which enabled the physiological role of glutamate to be established unequivocally, and also led to the realisation that EAA receptors are heterogeneous.
The realisation of glutamate’s importance came slowly (see Watkins & Jane, 2006). By the 1950s, work on the peripheral nervous system had highlighted the transmitter roles of acetylcholine and catecholamines, and as the brain also contained these substances, there seemed little reason to look further. The presence of γ-aminobutyric acid (GABA; see below) in the brain, and its powerful inhibitory effect on neurons, were discovered in the 1950s, and its transmitter role was postulated. At the same time, work by Curtis’s group in Canberra showed that glutamate and various other acidic amino acids produced a strong excitatory effect, but it seemed inconceivable that such workaday metabolites could actually be transmitters. Through the 1960s, GABA and excitatory amino acids (EAAs) were thought, even by their discoverers, to be mere pharmacological curiosities. In the 1970s, the humblest amino acid, glycine, was established as an inhibitory transmitter in the spinal cord, giving the lie to the idea that transmitters had to be exotic molecules, too beautiful for any role but to sink into the arms of a receptor. Once glycine had been accepted, the rest quickly followed. A major advance was the discovery of EAA antagonists, based on the work of Watkins in Bristol, which enabled the physiological role of glutamate to be established unequivocally, and also led to the realisation that EAA receptors are heterogeneous.
To do justice to the wealth of discovery in this field in the past two decades is beyond the range of this book; for more detail see Gereau & Swanson (2008). Here we concentrate on pharmacological aspects. After several beautiful but false dawns, a number of new drugs are in development on the basis of EAA mechanisms.1 The major problem has been that EAA-mediated neurotransmission is ubiquitous in the brain and so agonist and antagonist drugs exert effects at many sites, giving rise not only to therapeutically beneficial effects, but also to other, unwanted harmful effects.
Metabolism and Release of Amino Acids
Glutamate is widely and fairly uniformly distributed in the CNS, where its concentration is much higher than in other tissues. It has an important metabolic role, the metabolic and neurotransmitter pools being linked by transaminase enzymes that catalyse the interconversion of glutamate and α-oxoglutarate (Fig. 37.1). Glutamate in the CNS comes mainly from either glucose, via the Krebs cycle, or glutamine, which is synthesised by glial cells and taken up by the neurons; very little comes from the periphery. The interconnection between the pathways for the synthesis of EAAs and inhibitory amino acids (GABA and glycine), shown in Figure 37.1, makes it difficult to use experimental manipulations of transmitter synthesis to study the functional role of individual amino acids, because disturbance of any one step will affect both excitatory and inhibitory mediators.
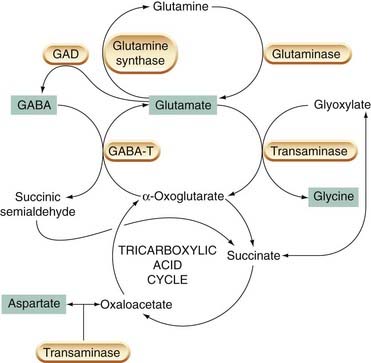
Fig. 37.1 Metabolism of transmitter amino acids in the brain.
Transmitter substances are marked with green boxes. GABA-T, GABA transaminase; GAD, glutamic acid decarboxylase.
In common with other fast neurotransmitters, glutamate is stored in synaptic vesicles and released by Ca2+-dependent exocytosis; specific transporter proteins account for its uptake by neurons and other cells, and for its accumulation by synaptic vesicles (see Ch. 12). Released glutamate is taken up into cells by Na+/H+/K+ dependent transporters (cf. monoamine transporters—Chs 12 & 14), and transported into synaptic vesicles, by a different transporter driven by the proton gradient across the vesicle membrane. Several EAA transporters have been cloned and characterised in detail (see Shigeri et al., 2004). There may be value in developing enhancers and inhibitors of glutamate uptake (see Bunch et al., 2009) for the treatment of CNS disorders in which the level of extracellular glutamate may be abnormal, e.g. neurodegeneration (see Ch. 39), schizophrenia (see Ch. 45) and depression (see Ch. 46). In contrast to the situation with monoamine synthesis and transport (Chs 14 and 38), few drugs (none in clinical use) are known that interfere specifically with glutamate metabolism.
The action of glutamate is terminated mainly by carrier-mediated reuptake into the nerve terminals and neighbouring astrocytes (Fig. 37.2). This transport can, under some circumstances (e.g. depolarisation by increased extracellular [K+]), operate in reverse and constitute a source of glutamate release (see Takahashi et al., 1997), a process that may occur under pathological conditions such as brain ischaemia (see Ch. 39). Glutamate taken up by astrocytes is converted to glutamine and recycled, via transporters, back to the neurons, which convert the glutamine back to glutamate. Glutamine, which lacks the pharmacological activity of glutamate, thus serves as a pool of inactive transmitter under the regulatory control of the astrocytes, which act as ball boys, returning the ammunition in harmless form in order to rearm the neurons.
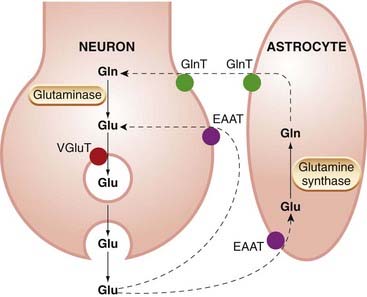
Fig. 37.2 Transport of glutamate (Glu) and glutamine (Gln) by neurons and astrocytes.
Released glutamate is captured partly by neurons and partly by astrocytes, which convert most of it to glutamine. EAAT, excitatory amino acid transporter; GlnT, glutamine transporter; VGluT, vesicular glutamate transporter.
Glutamate
Glutamate Receptor Subtypes
Glutamate and related excitatory amino acids activate both ionotropic (ligand-gated cation channels) and metabotropic (G-protein-coupled) receptors (see Ch. 3 for a general description of ionotropic and metabotropic receptors).
Ionotropic Glutamate Receptors
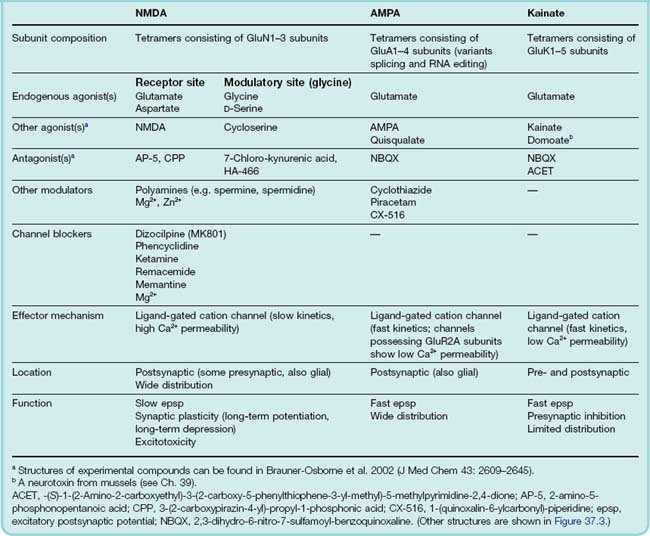
On the basis of studies with selective agonists and antagonists (Fig. 37.3), three main subtypes of ionotropic receptors for glutamate can be distinguished: NMDA, AMPA and kainate2 receptors, named originally according to their specific agonists (Table 37.1). These ligand-gated channels can be homomeric or heteromeric assemblies of four subunits, each with the ‘pore loop’ structure shown in Figure 3.18. There are some 16 different receptor subunits and their nomenclature has, until recently, been somewhat confusing.3 Here, in this brief, general description, we use the new International Union of Basic and Clinical Pharmacology (IUPHAR) recommended terminology because it simplifies the subject considerably, but beware confusion when reading older papers. NMDA receptors are assembled from seven types of subunit (GluN1, GluN2A, GluN2B, GluN2C, GluN2D, GluN3A, GluN3B). The subunits comprising AMPA receptors (GluA1–4)4 and kainate receptors (GluK1–5), are closely related to, but distinct from, GluN subunits. Receptors comprising different subunits can have different pharmacological and physiological characteristics, e.g. AMPA receptors lacking the GluA2 subunit have much higher permeability to Ca2+ than the others, which has important functional consequences (see Ch. 4).
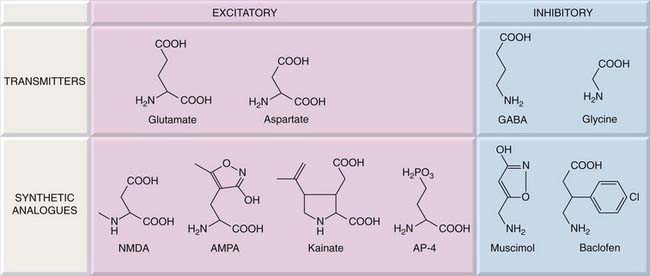
Fig. 37.3 Structures of agonists acting on glutamate, GABA and glycine receptors.
The receptor specificity of these compounds is shown in Tables 37.1 and 37.2. AMPA, α-amino-3-hydroxy-5-methylisoxazole-4-propianic acid; AP-4, 3-amino-4-phosphonopentanoic acid; NMDA, N-methyl-D-asparatic acid.
AMPA receptors, and in certain brain regions kainate receptors (see Bleakman & Lodge, 1998), serve to mediate fast excitatory synaptic transmission in the CNS—absolutely essential for our brains to function. Kainate and NMDA receptors are also expressed on nerve terminals where they can enhance or reduce transmitter release (see Corlew et al., 2008; Jane et al., 2009).5 AMPA receptors occur on astrocytes as well as on neurons, and these cells play an important role in communication in the brain. Postsynaptic NMDA receptors (which often coexist with AMPA receptors) contribute a slow component to the excitatory synaptic potential (Fig. 37.4), the magnitude of which varies in different pathways.
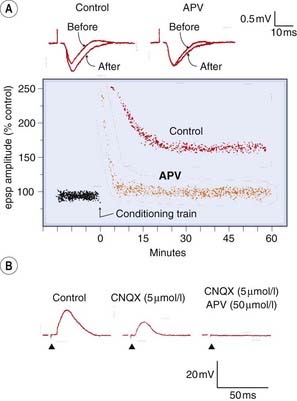
Fig. 37.4 Effects of excitatory amino acid receptor antagonists on synaptic transmission.
[A] APV (NMDA antagonist) prevents long-term potentiation (LTP) in the rat hippocampus without affecting the fast excitatory postsynaptic potential (epsp). Top records show the extracellularly recorded fast epsp (downward deflection) before, and 50 min after, a conditioning train of stimuli (100 Hz for 2 s). The presence of LTP in the control preparation is indicated by the increase in epsp amplitude. In the presence of APV (50 µmol/l), the normal epsp is unchanged, but LTP does not occur. Lower trace shows epsp amplitude as a function of time. The conditioning train produces a short-lasting increase in epsp amplitude, which still occurs in the presence of APV, but the long-lasting effect is prevented. [B] Block of fast and slow components of epsp by CNQX (6-cyano-7-nitroquinoxaline-2,3-dione; AMPA receptor antagonist) and APV (NMDA receptor antagonist). The epsp (upward deflection) in a hippocampal neuron recorded with intracellular electrode is partly blocked by CNQX (5 µmol/l), leaving behind a slow component, which is blocked by APV (50 µmol/l).
(From: [A] Malinow R, Madison D, Tsien R W 1988 Nature 335: 821; [B] Andreasen M, Lambert J D, Jensen M S 1989 J Physiol 414: 317–336.)
Binding studies show that ionotropic glutamate receptors are most abundant in the cortex, basal ganglia and sensory pathways. NMDA and AMPA receptors are generally co-localised, but kainate receptors have a much more restricted distribution. Expression of the many different receptor subtypes in the brain also shows distinct regional differences, but we have hardly begun to understand the significance of this extreme organisational complexity.
Special features of NMDA receptors
NMDA receptors and their associated channels have been studied in more detail than the other types and show special pharmacological properties, summarised in Fig. 37.5, which are postulated to play a role in pathophysiological mechanisms.
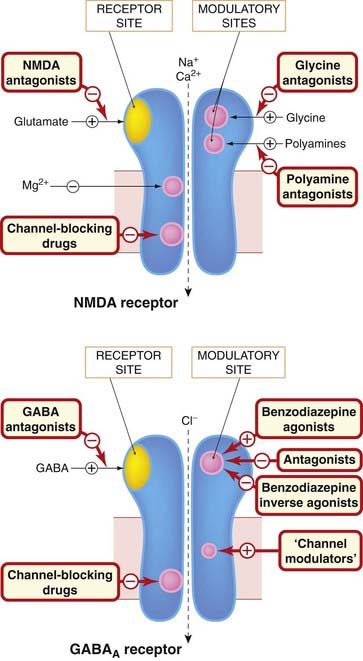
Fig. 37.5 Main sites of drug action on NMDA and GABAA receptors.
Both receptors are multimeric ligand-gated ion channels. Drugs can act as agonists or antagonists at the neurotransmitter receptor site or at modulatory sites associated with the receptor. They can also act to block the ion channel at one or more distinct sites. In the case of the GABAA receptor, the mechanism by which ‘channel modulators’ (e.g. ethanol, anaesthetic agents) facilitate channel opening is uncertain; they may affect both ligand binding and channel sites. The location of the different binding sites shown in the figure is largely imaginary, although study of mutated receptors is beginning to reveal where they actually reside. Examples of the different drug classes are given in Tables 37.1 and 37.3.
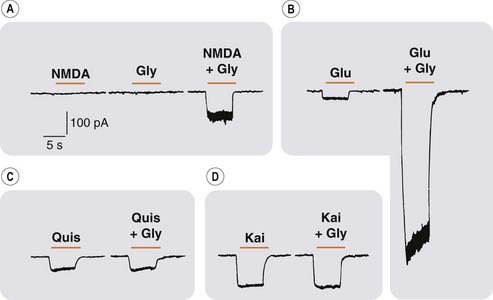
Fig. 37.6 Facilitation of NMDA by glycine.
Recordings from mouse brain neurons in culture (whole-cell patch clamp technique). Downward deflections represent inward current through excitatory amino acid-activated ion channels. [A] NMDA (10 µmol/l) or glycine (1 µmol/l) applied separately had little or no effect, but together produced a response. [B] The response to glutamate (Glu,10 µmol/l) was strongly potentiated by glycine (Gly, 1 µmol/l). [C] and [D] Responses of AMPA and kainate receptors to quisqualate (Quis) and kainate (Kai) were unaffected by glycine.
(From Johnson J W, Ascher P 1987 Nature 325: 529–531.)
Metabotropic Glutamate Receptors
There are eight different metabotropic glutamate receptors (mGlu1–8) which are unusual in showing no sequence homology with other G-protein-coupled receptors (Ferraguti & Shigemoto, 2006). They function as homodimers (see Ch. 3) cross-linked by a disulfide bridge across the extracellular domain of each protein (see Goudet et al., 2009). They are members of class C G-protein-coupled receptors, possessing a large extracellular N terminus domain that forms a venus fly trap-like structure into which glutamate binds. They can be divided into three groups on the basis of their sequence homology, G-protein coupling and pharmacology (see Table 37.2). Alternatively spliced receptor variants have been reported.
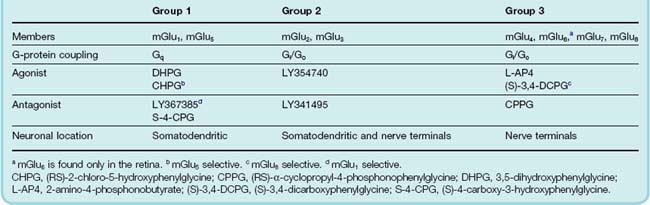
mGlu receptors are widely distributed throughout the central nervous system (see Ferraguti & Shigemoto, 2006) on neurons, where they regulate cell excitability and synaptic transmission, and on glia. Neuronal group 1 mGlu receptors are located postsynaptically and are largely excitatory. By raising intracellular [Ca2+], they modify responses through ionotropic glutamate receptors (see Fig. 37.7). Group 2 and 3 mGlu receptors are mostly presynaptic receptors and their activation tends to reduce synaptic transmission and neuronal excitability. They can be autoreceptors, involved in reducing glutamate release or heteroreceptors, e.g. when present on GABA-containing terminals.
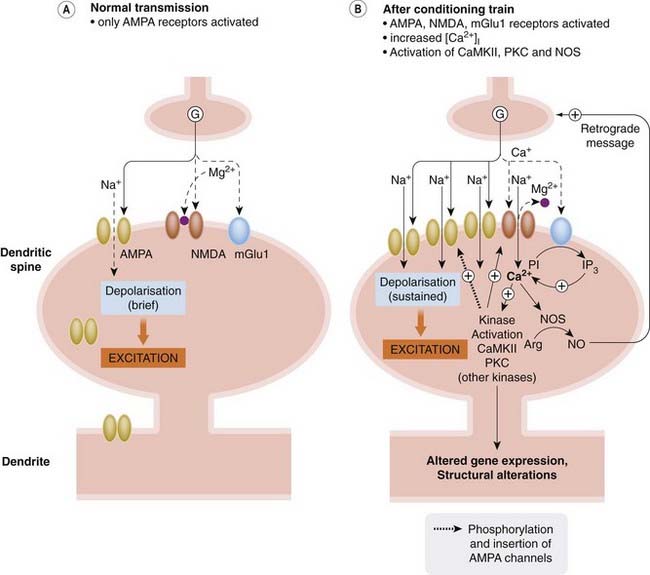
Fig. 37.7 Mechanisms of long-term potentiation.
[A] With infrequent synaptic activity, glutamate (G) activates mainly AMPA receptors. There is insufficient glutamate to activate metabotropic receptors, and NMDA receptor channels are blocked by Mg2+. [B] After a conditioning train of stimuli, enough glutamate is released to activate metabotropic receptors, and NMDA channels are unblocked by the sustained depolarisation. The resulting increase in [Ca2+]i activates various enzymes, including the following:
Arg, arginine; IP3, inositol (1,4,5) trisphosphate; NO, nitric oxide; PI, phosphatidylinositol.
Synaptic Plasticity and Long-Term Potentiation
In general, it appears that NMDA and mGlu receptors play a particular role in long-term adaptive and pathological changes in the brain, and are of particular interest as potential drug targets. AMPA receptors, on the other hand, are mainly responsible for fast excitatory transmission, and if they are fully blocked, brain function shuts down entirely; nevertheless, they too are involved in synaptic plasticity.
Two aspects of glutamate receptor function are of particular pathophysiological importance, namely synaptic plasticity, discussed here, and excitotoxicity (discussed in Ch. 39).
Synaptic plasticity is a general term used to describe long-term changes in synaptic connectivity and efficacy, either following physiological alterations in neuronal activity (as in learning and memory), or resulting from pathological disturbances (as in epilepsy, chronic pain or drug dependence). Synaptic plasticity underlies much of what we call ‘brain function’. Needless to say, no single mechanism is responsible; however, one significant and much-studied component is long-term potentiation (LTP), a phenomenon in which AMPA and NMDA receptors play a central role.
Long-term potentiation (LTP; see Bennett, 2000; Bear et al., 2006) is a prolonged (hours in vitro, days or weeks in vivo) enhancement of synaptic transmission that occurs at various CNS synapses following a short (conditioning) burst of high-frequency presynaptic stimulation. Its counterpart is long-term depression (LTD), which is produced at some synapses by a longer train of stimuli at lower frequency (see Massey & Bashir, 2007). These phenomena have been studied at various synapses in the CNS, most especially in the hippocampus which plays a central role in learning and memory (Fig. 37.4). It has been argued that ‘learning’, in the synaptic sense, can occur if synaptic strength is enhanced following simultaneous activity in both pre- and postsynaptic neurons. LTP shows this characteristic; it does not occur if presynaptic activity fails to excite the postsynaptic neuron, or if the latter is activated independently, for instance by a different presynaptic input. The mechanisms underlying both LTP and LTD differ somewhat at different synapses in the brain (see Bear et al., 2006). Here only a brief, generic view of the underlying events is given. LTP initiation may involve both presynaptic and postsynaptic components, and results from enhanced activation of postsynaptic AMPA receptors at EAA synapses and (probably) to enhanced glutamate release (although the argument rumbles on about whether increased transmitter release does or does not occur in LTP; see Blundon & Zakharenko, 2008). The response of postsynaptic AMPA receptors to glutamate is increased due to phosphorylation of the AMPA receptor subunits by kinases such as Ca2+/calmodulin-dependent protein kinase (CaMKII) and protein kinase C (PKC), thus enhancing their conductance, as well as to increased expression and trafficking of AMPA receptors to synaptic sites. LTD, on the other hand, results from modest Ca2+ entry into the cell through AMPA receptors (NMDA receptors remain blocked by Mg2+) activating phosphatases that reduce AMPA receptor phosphorylation and insertion into the plasma membrane.
LTP is reduced by agents that block the synthesis or effects of nitric oxide or arachidonic acid. These mediators (see Chs 17 and 20) may act as retrograde messengers through which events in the postsynaptic cell are able to influence the presynaptic nerve terminal. Anandamide, released by the postsynaptic cell, may also play a role by reducing the release of GABA from inhibitory nerve endings (see Ch. 18).
Two special properties of the NMDA receptor underlie its involvement in LTP, namely voltage-dependent channel block by Mg2+ and its high Ca2+ permeability. At normal membrane potentials, the NMDA channel is blocked by Mg2+; a sustained postsynaptic depolarisation produced by glutamate acting repeatedly on AMPA receptors, however, removes the Mg2+ block, and NMDA receptor activation then allows Ca2+ to enter the cell. Activation of group 1 mGlu receptors also contributes to the increase in [Ca2+]i. This rise in [Ca2+ ]i in the postsynaptic cell activates protein kinases, phospholipases and nitric oxide synthase, which act jointly with other cellular proceses (by mechanisms that are not yet fully understood) to facilitate transmission via AMPA receptors. Initially, during the induction phase of LTP, phosphorylation of AMPA receptors increases their responsiveness to glutamate. Later, during the maintenance phase, more AMPA receptors are recruited to the membrane of postsynaptic dendritic spines as a result of altered receptor trafficking; later still, various other mediators and signalling pathways are activated, causing structural changes and leading to a permanent increase in the number of synaptic contacts.
The general description of LTP given above is intended to provide the uninitiated reader with an overview of the topic. There are subtle differences in its forms and in the mechanisms underlying it at different synapses in the CNS. How LTP, in all of its guises, relates to different forms of memory is slowly being worked out (see Bear et al., 2006; Kessels & Malinow, 2009). Thus there is hope that drugs capable of enhancing LTP may improve learning and memory.
Drugs Acting on Glutamate Receptors
Antagonists and Negative Modulators
Inotropic glutamate receptor antagonists
The main types and examples of ionotropic glutamate antagonists are shown in Table 37.1. They are selective for the main receptor types but generally not for specific subtypes. Many of these compounds, although very useful as experimental tools in vitro, are unable to penetrate the blood–brain barrier, so they are not effective when given systemically.
NMDA receptors, as discussed above, require glycine as well as NMDA to activate them, so blocking of the glycine site is an alternative way to produce antagonism. Kynurenic acid and the more potent analogue 7-chloro-kynurenic acid act in this way, as do various compounds currently in development. Another site of block is the channel itself, where various substances act, for example ketamine and phencyclidine. Dizocilpine, remacemide and memantine are more recent examples. These agents are lipid soluble and thus able to cross the blood–brain barrier.
The potential therapeutic interest in ionotropic glutamate receptor antagonists lies mainly in the reduction of brain damage following strokes and head injury (Ch. 39), as well as in the treatment of epilepsy (Ch. 44) and Alzheimer’s disease (Ch. 39). They have also been considered for indications such as drug dependence (Ch. 48) and schizophrenia (Ch. 45). Trials with NMDA antagonists and channel blockers have so far proved disappointing, and a serious drawback of these agents is their tendency to cause hallucinatory and other disturbances (also a feature of phencyclidine; Ch. 47). Only two NMDA receptor antagonists, ketamine (anaesthesia and analgesia; see Chs 40 and 41) and memantine (Alzheimer’s disease; Ch. 39), are in clinical use. It is possible that antagonists selective for NMDA receptors containing the GluN2B subunit, which is highly Ca2+ permeable, may be effective for treating neurodegeneration and have fewer CNS side effects. Glycine site antagonists may also have fewer unwanted effects, and experimental compounds have been tested in clinical trials for conditions such as stroke and epilepsy (see Jansen & Dannhart, 2003); the results have been inconclusive. AMPA receptor antagonists seem unpromising as therapeutic agents, because the available agents (as might be expected) produce overall CNS depression, including respiratory depression, cognition impairment and motor incoordination, with little margin of safety. Only if subtype selectivity can be achieved is this approach likely to succeed. The prospects for kainate receptor antagonists appear more promising—antagonists for GluK1 have shown potential for the treatment of pain, migraine, epilepsy, stroke and anxiety (see Jane et al., 2009).
Overall, the promise foreseen for ionotropic glutamate receptor antagonists in the clinic has simply not, so far, been fulfilled. The problem may be that glutamate is such a ubiquitous and multifunctional mediator—involved, it seems, in almost every aspect of brain function—that attempting to improve a specific malfunction by flooding the brain with a compound that affects the glutamate system in some way is just too crude a strategy.
Metabotropic glutamate receptor antagonists
While antagonists that discriminate between the different groups of mGlu receptors are available (see Table 37.2), it has proven more difficult to develop selective antagonists for the subtypes within the groups. mGlu receptors, like many G-protein-coupled receptors, possess allosteric modulatory sites (see Ch. 3). Allosteric modulation can be either inhibitory or facilitatory. Antagonists or negative modulators acting at group 1 mGlu receptors have potential for the treatment of various pain states, Parkinson’s disease, neuroprotection, epilepsy and drug abuse; whereas antagonists or negative modulators of group 2 mGlu receptors have potential as cognition enhancers (Kew, 2004).
Agonists and Positive Modulators
Ionotropic glutamate receptors
Various agonists at ionotropic glutamate receptors that are used experimentally are shown in Table 37.1. From the clinical perspective, interest centres on the theory that positive AMPA receptor modulators may improve memory and cognitive performance. Examples include cyclothiazide, piracetam and CX-516 (Ampalex). These positive modulators, known as ampakines, are allosteric modulators and can act in subtly different ways to increase response amplitude, slow deactivation and attenuate desensitisation of AMPA receptor-mediated currents. They therefore increase AMPA-mediated synaptic responses and enhance long-term potentiation as well as upregulating the production of nerve growth factors such as brain-derived neurotrophic factor (BDNF). They are thought to have therapeutic potential as cognition enhancers and in the treatment of schizophrenia, depression, attention deficit hyperactivity disorder (ADHD) and Parkinson’s disease (see Lynch, 2006).
Metabotropic glutamate receptors
Agonists at group 2 and 3 mGlu receptors decrease glutamate release. They therefore have therapeutic potential to decrease neuronal cell death in stroke and in the treatment of epilepsy and anxiety as well as in controlling the positive symptoms of schizophrenia. As with antagonists (see above), developing selective agonists of mGlu receptors has proven to be quite difficult; the hope is that it will be easier to develop highly selective positive allosteric modulators (see Kew, 2004).
Excitatory amino acids 
γ-Aminobutyric Acid
GABA is the main inhibitory transmitter in the brain. In the spinal cord and brain stem, glycine is also important.
Synthesis, Storage and Function
GABA occurs in brain tissue but not in other mammalian tissues, except in trace amounts. It is particularly abundant (about 10 µmol/g tissue) in the nigrostriatal system, but occurs at lower concentrations (2–5 µmol/g) throughout the grey matter.
GABA is formed from glutamate (Fig. 37.1) by the action of glutamic acid decarboxylase (GAD), an enzyme found only in GABA-synthesising neurons in the brain. Immunohistochemical labelling of GAD is used to map the GABA pathways in the brain. GABAergic neurons and astrocytes take up GABA via specific transporters, thus removing GABA after it has been released. GABA transport is inhibited by guvacine, nipecotic acid and tiagabine. Tiagabine is used to treat epilepsy (Ch. 44). GABA can be destroyed by a transamination reaction in which the amino group is transferred to α-oxoglutaric acid (to yield glutamate), with the production of succinic semialdehyde and then succinic acid. This reaction is catalysed by GABA transaminase, an enzyme located primarily in astrocytes. It is inhibited by vigabatrine, another compound used to treat epilepsy (Ch. 44).
GABA functions as an inhibitory transmitter in many different CNS pathways. About 20% of CNS neurons are GABAergic; most are short interneurons, but there are some long GABAergic tracts, e.g. from the striatum to the substantia nigra and globus pallidus (see Ch. 39 and Fig. 39.4). The widespread distribution of GABA—GABA serves as a transmitter at about 30% of all the synapses in the CNS—and the fact that virtually all neurons are sensitive to its inhibitory effect suggests that its function is ubiquitous in the brain. That antagonists such as bicuculline (see below) induce seizures illustrates the important, ongoing inhibitory role of GABA in the brain.
Gaba Receptors: Structure and Pharmacology
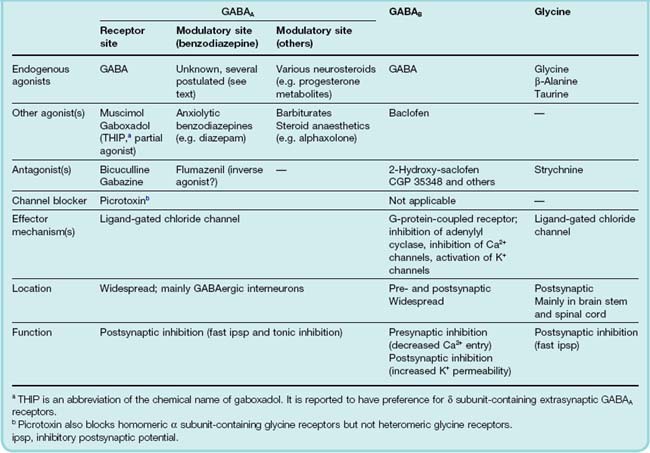
GABA acts on two distinct types of receptor: GABAA receptors are ligand-gated ion channels whereas the other, GABAB receptors, are G-protein coupled.
Gabaa Receptors
GABAA receptors7 (see Barnard, 2000) are members of the Cys loop family of receptors that also includes the glycine, nicotinic, and 5-HT3 receptors (see Fig. 3.18). The GABAA receptors are pentamers made up of different subunits. The reader should not despair when informed that nineteen GABAA receptor subunits have been cloned (α1–6, β1–3, γ1–3, δ, ε, θ, π and ρ1–3) and that splice variants of some subunits also exist. Although the number of possible combinations is large, only a few dozen have been shown to exist (Mody & Pearce, 2004). The most common are α1β2γ2 (by far the most abundant), α2β3γ2 and α3β1–3γ2 subunits. To make up the pentamer, each receptor contains 2 α, 2 β and 1 γ subunit arranged in a circle in the sequence α–β–α–β–γ around the pore when viewed from the extracellular side of the membrane. GABA binds at the interface between the α and β subunits whereas benzodiazepines (see Ch. 43) bind at the α/γ interface. Receptors containing different α and γ subunits exhibit differential sensitivity to benzodiazepines and mediate different behavioural responses to these drugs. This raises the tantalising prospect of developing new agents with greater selectivity and potentially fewer side effects. The GABAA receptor should therefore be thought of as a group of receptors exhibiting subtle differences in their physiological and pharmacological properties.
GABAA receptors are primarily located postsynaptically and mediate fast postsynaptic inhibition, the channel being selectively permeable to Cl−. Because the equilibrium membrane potential for Cl− is usually negative to the resting potential, increasing Cl− permeability hyperpolarises the cell as Cl− ions enter, thereby reducing its excitability.8 GABAA receptors are located both at areas of synaptic contact and extrasynaptically (Farrant & Nusser, 2005). Thus GABA produces inhibition by acting both as a fast ‘point-to-point’ transmitter and as an ‘action-at-a-distance’ neuromodulator, as the extrasynaptic GABAA receptors can be tonically activated by GABA that has diffused away from its site of release. Extrasynaptic GABAA receptors contain α4, α5 and α6 subunits as well as the δ subunit, and are highly sensitive to general anaesthetic agents (see Ch. 40) and ethanol (see Ch. 48), have higher affinities for GABA and show less desensitisation. Gaboxadol (previously known as THIP from its chemical structure) is a selective GABAA receptor agonist with a preference for δ subunit-containing GABAA receptors.
Gabab Receptors
GABAB receptors (see Bettler et al., 2004) are located pre- and postsynaptically. They are class C G-protein-coupled receptors that couple through Gi/Go to inhibit voltage-gated Ca2+ channels (thus reducing transmitter release), to open potassium channels (thus reducing postsynaptic excitability) and to inhibit adenylyl cyclase.
For GABAB receptors, the functional receptor is a dimer (see Ch. 3) consisting of two different seven-transmembrane subunits, B1 and B2, held together by a coil/coil interaction between their C-terminal tails (Kubo & Tateyama, 2005). In the absence of B2, the B1 subunit does not traffic to the plasma membrane as it possesses an endoplasmic reticulum retention signal. Interaction of B1 with B2 masks the retention signal and facilitates trafficking to the membrane. Activation of the dimer results from GABA binding to the extracellular, venus fly trap-like domain of B1 (even although the B2 subunit possesses a similar domain) whereas it is the B2 subunit that interacts with and activates the G-protein (Fig. 37.8).
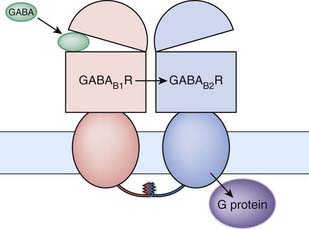
Fig 37.8 Dimeric structure of the GABAB receptor.
The receptor is made up of two seven-transmembrane domain subunits held together by a coil/coil interaction between their C-terminal tails. Activation of the receptor occurs when GABA binds to the extracellular domain of the B1 subunit. This produces an allosteric change in the B2 subunit which is coupled to the G-protein.
(Adapted from Kubo & Tateyama, 2005 Current Opinion in Neurobiology. 15: 289–295.)
Drugs Acting on Gaba Receptors
Gabaa Receptors
GABAA receptors resemble NMDA receptors in that drugs may act at several different sites (Fig. 37.5; see Johnston, 1996). These include:
There is growing evidence that the different receptor subtypes differ in their pharmacological properties.
GABAA receptors are the target for several important centrally acting drugs, notably benzodiazepines, barbiturates, neurosteroids (see below) and several general anaesthetics. The main agonists, antagonists and modulatory substances that act on GABA receptors are shown in Table 37.3.
Muscimol, derived from a hallucinogenic mushroom, resembles GABA chemically and is a powerful GABAA receptor agonist. A synthetic analogue, gaboxadol is a partial agonist that was developed as a hypnotic drug (Ch. 43) but has now been withdrawn. Bicuculline, a naturally occurring convulsant compound, is a specific antagonist that blocks the fast inhibitory synaptic potential in most CNS synapses. Gabazine, a synthetic GABA analogue, is similar. These compounds are useful experimental tools but have no therapeutic uses.
Benzodiazepines, which have powerful sedative, anxiolytic and anticonvulsant effects (see Ch. 43), selectively potentiate the effects of GABA on some GABAA receptors depending upon the subunit composition of the receptor. They bind with high affinity to an accessory site (the ‘benzodiazepine receptor’) on the GABAA receptor, in such a way that the binding of GABA is facilitated and its agonist effect is enhanced. Conversely, inverse agonists at the benzodiazepine receptor (e.g. Ro15-4513) reduce GABA binding and are anxiogenic and proconvulsant—they are unlikely to be therapeutically useful!
Modulators that also enhance the action of GABA, but whose site of action is less well defined than that of benzodiazepines (shown as ‘channel modulators’ in Fig. 37.5), include other CNS depressants such as barbiturates (Ch. 43), anaesthetic agents (Ch. 40) and neurosteroids. Neurosteroids (see Lambert et al., 2003) are compounds that are related to steroid hormones but that act (like benzodiazepines) to enhance activation of GABAA receptors as well as on conventional intracellular steroid receptors. Interestingly, they include metabolites of progesterone and androgens that are formed in the nervous system, and are believed to have a physiological role. Synthetic neurosteroids include alphaxolone, developed as an anaesthetic agent (Ch. 40).
Picrotoxin is a convulsant that acts by blocking the chloride channel associated with the GABAA receptor, thus blocking the postsynaptic inhibitory effect of GABA. It has no therapeutic uses.
Gabab Receptors
When the importance of GABA as an inhibitory transmitter was recognised, it was thought that a GABA-like substance might prove to be effective in controlling epilepsy and other convulsive states; because GABA itself fails to penetrate the blood–brain barrier, more lipophilic GABA analogues were sought, one of which, baclofen (see Fig. 37.3), was introduced in 1972. Unlike GABA, its actions are not blocked by bicuculline. These findings led to the recognition of the GABAB receptor, for which baclofen is a selective agonist (see Bowery, 1993). Baclofen is used to treat spasticity and related motor disorders (Ch. 44) and may also be useful in the treatment of drug dependence (see Ch. 48).
Competitive antagonists for the GABAB receptor include a number of experimental compounds (e.g. 2-hydroxy-saclofen and more potent compounds with improved brain penetration, such as CGP 35348). Tests in animals have shown that these compounds produce only slight effects on CNS function (in contrast to the powerful convulsant effects of GABAA antagonists). The main effect observed, paradoxically, was an antiepileptic action, specifically in an animal model of absence seizures (see Ch. 44), together with enhanced cognitive performance. Whether such compounds will prove to have therapeutic uses remains to be seen.
γ-Hydroxybutyrate
γ-Hydroxybutyrate (GHB; see Wong et al., 2004) occurs naturally in the brain as a side product of GABA synthesis. As a synthetic drug from 1960 onwards, it has found favour with bodybuilders, based on its ability to evoke the release of growth hormone, and with party-goers, based on its euphoric and disinhibitory effects. In common with many abused drugs (see Ch. 48), it activates ‘reward pathways’ in the brain, and its use is now illegal in most countries. The pharmacological properties of GHB are not well understood, although it is believed to be a weak partial agonist at GABAB receptors and to bind to specific GHB receptor sites (see Wu et al., 2004), of which little is known.
Glycine
Glycine is present in particularly high concentration (5 µmol/g) in the grey matter of the spinal cord. Applied ionophoretically to motor neurons or interneurons, it produces an inhibitory hyperpolarisation that is indistinguishable from the inhibitory synaptic response. Strychnine, a convulsant poison that acts mainly on the spinal cord, blocks both the synaptic inhibitory response and the response to glycine. This, together with direct measurements of glycine release in response to nerve stimulation, provides strong evidence for its physiological transmitter role. β-Alanine has pharmacological effects and a pattern of distribution very similar to those of glycine, but its action is not blocked by strychnine.
The inhibitory effect of glycine is quite distinct from its role in facilitating activation of NMDA receptors (see p. 450).
The glycine receptor (see Lynch, 2009) resembles the GABAA receptor in that it is a Cys loop, pentameric ligand-gated chloride channel. There are no specific metabotropic receptors for glycine. Five glycine receptor subunits have been cloned (α1–4, β) and it appears that in the adult brain the main form of receptor is made up of α1 and β subunits, although debate is ongoing about the exact stoichiometry. The situation for glycine is therefore much simpler than for GABA (see above). Mutations of the receptor have been identified in some inherited neurological disorders associated with muscle spasm and reflex hyperexcitability. There are no therapeutic drugs that act specifically by modifying glycine receptors, although it turns out that many of the compounds (such as benzodiazepines and anaesthetic agents) that enhance GABAA receptor activation act similarly on glycine receptors.
Tetanus toxin, a bacterial toxin resembling botulinum toxin (Ch. 13), acts selectively to prevent glycine release from inhibitory interneurons in the spinal cord, causing excessive reflex hyperexcitability and violent muscle spasms (lockjaw).
Glycine is removed from the extracellular space by two transporters GlyT1 and GlyT2 (Eulenburg et al., 2005). GlyT1 is located primarily on astrocytes and expressed throughout most regions of the CNS. GlyT2 on the other hand is expressed on glycinergic neurons in the spinal cord, brain stem and cerebellum. As described above, in addition to its function as an inhibitory transmitter, glycine also functions as a co-agonist with glutamate at NMDA receptors. Inhibition of glycine uptake by GlyT1 leads to an elevation of extracellular glycine levels throughout the brain and, through potentiation of NMDA receptor-mediated responses, could be beneficial in the treatment of schizophrenia (see Ch. 45). Another potential use of glycine transporter inhibitors could be as analgesics.