38 Other transmitters and modulators
Overview
The principal ‘amine’ transmitters in the central nervous system (CNS), namely noradrenaline, dopamine, 5-hydroxytryptamine (5-HT, serotonin) and acetylcholine (ACh), are described in this chapter, with briefer coverage of other mediators, including histamine, melatonin and purines. The monoamines were the first CNS transmitters to be identified, and during the 1960s a combination of neurochemistry and neuropharmacology led to many important discoveries about their role, and about the ability of drugs to influence these systems. Amine mediators differ from the amino acid transmitters discussed in Chapter 37 in being localised to small populations of neurons with cell bodies in the brain stem and basal forebrain, which project diffusely both rostrally to cortical and other areas, and in some cases caudally to the spinal cord. These amine-containing neurons are broadly associated with high-level behaviours (e.g. emotion, cognition and awareness), rather than with localised synaptic excitation or inhibition.1 More recently, some ‘atypical’ chemical mediators, such as nitric oxide (NO; Ch. 20) and endocannabinoids (Ch. 18) have come on the scene, and they are discussed at the end of the chapter. The other major class of CNS mediators, the neuropeptides, are described in Chapter 19, and information on specific neuropeptides (e.g. endorphins and neurokinins) appears in later chapters in this section.
Introduction
Although we know much about the many different mediators, their cognate receptors and signalling mechanisms at the cellular level, when describing their effects on brain function and behaviour we fall back on relatively crude terms—psychopharmacologists will be at our throats for so under-rating the sophistication of their measurements—such as ‘motor coordination’, ‘arousal’, ‘cognitive impairment’ and ‘exploratory behaviour’. The gap between these two levels of understanding still frustrates the best efforts to link drug action at the molecular level to drug action at the therapeutic level. Modern approaches, such as the use of transgenic animal technology (see Ch. 7) and non-invasive imaging techniques, are helping to forge links, but there is still a long way to go.
More detail on the content of this chapter can be found in Davis et al. (2002), Nestler et al. (2008) and Iversen et al. (2009).
Noradrenaline
The basic processes responsible for the synthesis, storage and release of noradrenaline are the same in the CNS as in the periphery (Ch. 14). In the CNS, inactivation of released noradrenaline is by neuronal reuptake or by metabolism, largely through the monamine oxidase, aldehyde reductase and catechol-O-methyl transferase mediated pathway to 3-hydroxy-4-methoxyphenylglycol (MHPG) (see Fig. 14.4).
Noradrenergic Pathways in the CNS
Although the transmitter role of noradrenaline in the brain was suspected in the 1950s, detailed analysis of its neuronal distribution became possible only when a technique, based on the formation of fluorescent catecholamine derivatives when tissues are exposed to formaldehyde, was devised by Falck and Hillarp. Detailed maps of the pathway of noradrenergic, dopaminergic and serotonergic neurons in laboratory animals were produced and later confirmed in human brains. The cell bodies of noradrenergic neurons occur in small clusters in the pons and medulla, and they send extensively branching axons to many other parts of the brain and spinal cord (Fig. 38.1). The most prominent cluster is the locus coeruleus (LC), located in the pons. Although it contains only about 10 000 neurons in humans, the axons, running in a discrete medial forebrain bundle, give rise to many millions of noradrenergic nerve terminals throughout the cortex, hippocampus, thalamus, hypothalamus and cerebellum. These nerve terminals do not form distinct synaptic contacts but appear to release transmitter somewhat diffusely. The LC also projects to the spinal cord and is involved in the descending control of pain (Ch. 41).
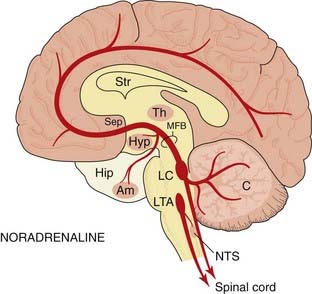
Fig. 38.1 Simplified diagram of the noradrenaline pathways in the brain.
The location of the main groups of cell bodies and fibre tracts is in solid colour. Light-shaded areas show the location of noradrenergic terminals. Am, amygdaloid nucleus; C, cerebellum; Hip, hippocampus; Hyp, hypothalamus; LC, locus coeruleus; LTA, lateral tegmental area, part of the reticular formation; MFB, medial forebrain bundle; NTS, nucleus of the tractus solitarius (vagal sensory nucleus); Sep, septum; Str, corpus striatum; Th, thalamus.
Other noradrenergic neurons lie close to the LC in the pons and project to the amygdala, hypothalamus, hippocampus and other parts of the forebrain, as well as to the spinal cord. A small cluster of adrenergic neurons, which release adrenaline rather than noradrenaline, lies more ventrally in the brain stem. These cells contain phenylethanolamine N-methyl transferase, the enzyme that converts noradrenaline to adrenaline (see Ch. 14), and project mainly to the pons, medulla and hypothalamus. Rather little is known about them, but they are believed to be important in cardiovascular control.
Functional Aspects
With the exception of the β3 adrenoceptor, all of the adrenoceptors (α1A, α1B, α1C, α2A, α2B, α2C, β1 and β2) are expressed in the CNS (see Bylund, 2007). They are G-protein-coupled receptors that interact with a variety of effector mechanisms (see Table 14.1). The role of α1 receptors in the CNS is poorly understood. They are widely distributed, located both on postsynaptic neurons and on glial cells, and may be involved in motor control, cognition and fear. α2 Adrenoceptors are located on noradrenergic neurons (in both somatodendritic and nerve terminal regions where they function as inhibitory autoreceptors) as well as on postsynaptic non-noradrenergic neurons. They are involved in blood pressure control (see below), sedation (α2 agonists such as medetomidine are used as anaesthetics in veterinary practice) and analgesia. β1 Receptors are found in the cortex, striatum and hippocampus whereas β2 receptors are largely found in the cerebellum. They have been implicated in the long-term effects of antidepressant drugs but quite how remains a mystery (see Ch. 46).
Research on the α2 adrenoceptor antagonist, idazoxan, has led to the identification of other putative imidazoline ‘receptors’ (see Head & Mayorov, 2006). These are the I1 receptor, which plays a role in the central control of blood pressure; the I2 receptor, an allosteric binding site on monoamine oxidase, and the I3 receptor, present in the pancreas with a role in regulating insulin secretion.
Arousal and mood
Attention has focused mainly on the LC, which is the source of most of the noradrenaline released in the brain, and from which neuronal activity can be measured by implanted electrodes. LC neurons are silent during sleep, and their activity increases with behavioural arousal. ‘Wake-up’ stimuli of an unfamiliar or threatening kind excite these neurons much more effectively than familiar stimuli. Amphetamine-like drugs, which release catecholamines in the brain, increase wakefulness, alertness and exploratory activity (although, in this case, firing of LC neurons is actually reduced by feedback mechanisms; see Ch. 47).
There is a close relationship between mood and state of arousal; depressed individuals are usually lethargic and unresponsive to external stimuli. The catecholamine hypothesis of depression (see Ch. 46) suggested that it results from a functional deficiency of noradrenaline in certain parts of the brain, while mania results from an excess. This remains controversial, and subsequent findings suggest that 5-HT may be more important than noradrenaline in relation to mood.
Blood pressure regulation
The role of central, as well as peripheral, noradrenergic synapses in blood pressure control is shown by the action of hypotensive drugs such as clonidine and methyldopa (see Chs 14 and 22) which decrease the discharge of sympathetic nerves emerging from the CNS. They cause hypotension when injected locally into the medulla or fourth ventricle, in much smaller amounts than are required when the drugs are given systemically. Noradrenaline and other α2 adrenoceptor agonists have the same effect when injected locally. Noradrenergic synapses in the medulla probably form part of the baroreceptor reflex pathway, because stimulation or antagonism of α2 adrenoceptors in this part of the brain has a powerful effect on the activity of baroreceptor reflexes.
Ascending noradrenergic fibres run to the hypothalamus, and descending fibres run to the lateral horn region of the spinal cord, acting to increase sympathetic discharge in the periphery. It has been suggested that these regulatory neurons may release adrenaline rather than noradrenaline as inhibition of phenylethanolamine N-methyl transferase, the enzyme that converts noradrenaline to adrenaline, interferes with the baroreceptor reflex.
Moxonidine, reported to be an I1 receptor agonist with less activity at α2 adrenoceptors, acts centrally to reduce peripheral sympathetic activity, thus decreasing peripheral vascular resistance.
Noradrenaline in the CNS 
Dopamine
Dopamine is particularly important in relation to neuropharmacology, because it is involved in several common disorders of brain function, notably Parkinson’s disease, schizophrenia and attention deficit disorder, as well as in drug dependence and certain endocrine disorders. Many of the drugs used clinically to treat these conditions work by influencing dopamine transmission.
The distribution of dopamine in the brain is more restricted than that of noradrenaline. Dopamine is most abundant in the corpus striatum, a part of the extrapyramidal motor system concerned with the coordination of movement (see Ch. 39), and high concentrations also occur in certain parts of the frontal cortex, limbic system and hypothalamus (where its release into the pituitary blood supply inhibits secretion of prolactin; Ch. 32).
The synthesis of dopamine follows the same route as that of noradrenaline (see Fig. 14.2), namely conversion of tyrosine to dopa (the rate-limiting step), followed by decarboxylation to form dopamine. Dopaminergic neurons lack dopamine β-hydroxylase, and thus do not convert dopamine to noradrenaline.
Dopamine is largely recaptured, following its release from nerve terminals, by a specific dopamine transporter, one of the large family of monoamine transporters (see Ch. 14). It is metabolised by monoamine oxidase and catechol-O-methyl transferase (Fig. 38.2), the main products being dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA, the methoxy derivative of DOPAC). The brain content of HVA is often used as an index of dopamine turnover. Drugs that cause the release of dopamine increase HVA, often without changing the concentration of dopamine. DOPAC and HVA, and their sulfate conjugates, are excreted in the urine, which provides an index of dopamine release in human subjects.
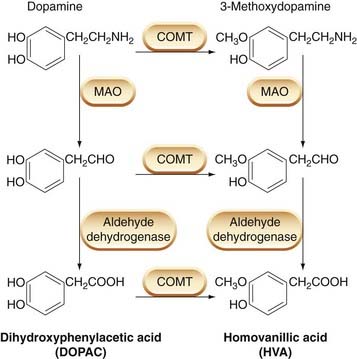
Fig. 38.2 The main pathways for dopamine metabolism in the brain.
COMT, catechol-O-methyl transferase; MAO, monoamine oxidase.
6-Hydroxydopamine, which selectively destroys dopaminergic nerve terminals, is commonly used as a research tool. It is taken up by the dopamine transporter and converted to a reactive metabolite that causes oxidative cytotoxicity.
Dopaminergic Pathways in the CNS
There are four main dopaminergic pathways in the brain (Fig. 38.3):
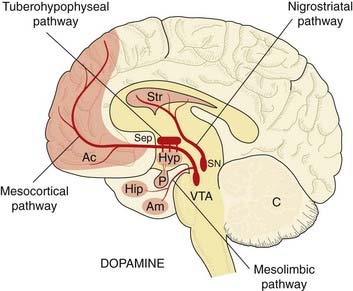
Fig. 38.3 Simplified diagram of the dopamine pathways in the brain, drawn as in Figure 38.1.
The pituitary gland (P) is shown, innervated with dopaminergic fibres from the hypothalamus. Ac, nucleus accumbens; SN, substantia nigra; VTA, ventral tegmental area; other abbreviations as in Figure 38.1.
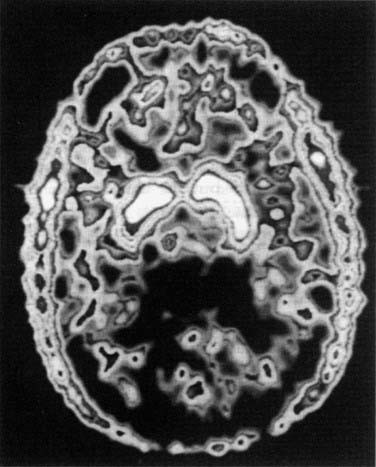
Fig. 38.4 Dopamine in the basal ganglia of a human subject.
The subject was injected with 5-fluoro-dopa labelled with the positron-emitting isotope 18F, which was localised 3 h later by the technique of positron emission tomography. The isotope is accumulated (white areas) by the dopa uptake system of the neurons of the basal ganglia, and to a smaller extent in the frontal cortex. It is also seen in the scalp and temporalis muscles.
(From Garnett E S et al. Nature 305: 137.)
There are also dopaminergic neurons in other brain regions and in the retina. For a more complete description, see Björklund & Dunnett (2007). The functions of the main dopaminergic pathways are discussed below.
Dopamine Receptors
Two types of receptor, D1 and D2, were originally distinguished on pharmacological and biochemical grounds. Gene cloning revealed further subgroups, D1 to D5 (for review, see Missale et al., 1998). The original D1 family now includes D1 and D5, while the D2 family, which is pharmacologically more important in the CNS, consists of D2, D3 and D4 (see Table 38.1). Splice variants, leading to long and short forms of D2, and genetic polymorphisms, particularly of D4 (see below), have subsequently been identified.
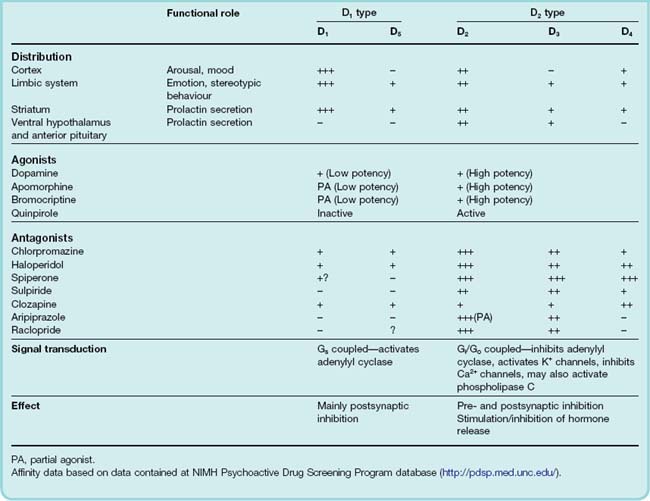
 All belong to the family of G-protein-coupled transmembrane receptors described in Chapter 3—D1 and D5 link through Gs to stimulate adenylyl cyclase; D2, D3, and D4 link through Gi/Go and activate potassium channels as well as inhibiting calcium channels and adenylyl cyclase. In addition they can also affect other cellular second messenger cascades (see Ch. 3). A key component in the signal transduction pathway is the protein DARPP-32 (32-kDa dopamine- and cAMP-regulated phosphoprotein; see Girault & Greengard, 2004). When intracellular cAMP is increased through activation of D1 receptors, activating protein kinase A, DARPP-32 is phosphorylated (Fig. 38.5). Phosphorylated DARPP-32 acts as an inhibitor of protein phosphatases such as protein phosphatase-1 and calcineurin, thus acting in concert with protein kinases and favouring protein phosphorylation—effectively an amplifying mechanism. In general, activation of D2 receptors opposes the effect of D1 receptor activation.
All belong to the family of G-protein-coupled transmembrane receptors described in Chapter 3—D1 and D5 link through Gs to stimulate adenylyl cyclase; D2, D3, and D4 link through Gi/Go and activate potassium channels as well as inhibiting calcium channels and adenylyl cyclase. In addition they can also affect other cellular second messenger cascades (see Ch. 3). A key component in the signal transduction pathway is the protein DARPP-32 (32-kDa dopamine- and cAMP-regulated phosphoprotein; see Girault & Greengard, 2004). When intracellular cAMP is increased through activation of D1 receptors, activating protein kinase A, DARPP-32 is phosphorylated (Fig. 38.5). Phosphorylated DARPP-32 acts as an inhibitor of protein phosphatases such as protein phosphatase-1 and calcineurin, thus acting in concert with protein kinases and favouring protein phosphorylation—effectively an amplifying mechanism. In general, activation of D2 receptors opposes the effect of D1 receptor activation.
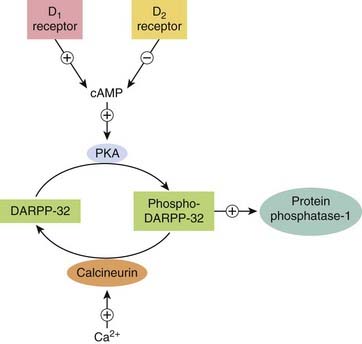
Fig. 38.5 The role of the neuron-specific phosphoprotein DARPP-32 in signalling by dopamine receptors (see text).
Dopamine receptors are expressed in the brain in distinct but overlapping areas. D1 receptors are the most abundant and widespread in areas receiving a dopaminergic innervation (namely the striatum, limbic system, thalamus and hypothalamus; Fig. 38.3), as are D2 receptors, which also occur in the pituitary gland. D2 receptors are found not only on dopaminergic neurons (cell bodies, dendrites and nerve terminals), where they function as inhibitory autoreceptors, but also on non-dopaminergic neurons (see De Mei et al., 2009). D3 receptors occur in the limbic system but not in the striatum. The D4 receptor is much more weakly expressed, mainly in the cortex and limbic systems.
The D4 receptor displays an unexpected polymorphism in humans, with a varying number (from 2 to 10) of 16 amino acid repeat sequences being expressed in the third intracellular loop, which participates in G-protein coupling (Ch. 3). Expectations that D4 receptor polymorphism might be related to the occurrence of schizophrenia in humans were disappointed after several studies failed to find any correlation (Tarazi et al., 2004). There may be a connection with attention deficit hyperactivity disorder (see Thapar et al., 2007).
Dopamine, like many other transmitters and modulators, acts presynaptically as well as postsynaptically. Presynaptic D2 receptors occur mainly on dopaminergic neurons, for example those in the striatum and limbic system, where they act to inhibit dopamine synthesis and release. Dopamine antagonists, by blocking these receptors, increase dopamine synthesis and release, and cause accumulation of dopamine metabolites in these parts of the brain. They also cause an increase in the rate of firing of dopaminergic neurons, probably by blocking feedback at the somatodendritic level mediated by locally released dopamine.
Dopamine receptors also mediate various effects in the periphery (mediated by D1 receptors), notably renal vasodilatation and increased myocardial contractility (dopamine itself has been used clinically in the treatment of circulatory shock; see Ch. 21).
Functional Aspects
The functions of dopaminergic pathways divide broadly into:
Dopamine and motor systems
Ungerstedt showed, in 1968, that bilateral ablation of the substantia nigra in rats, which destroys the nigrostriatal neurons, causes profound catalepsy, the animals becoming so inactive that they die of starvation unless artificially fed. Parkinson’s disease (Ch. 39) is a disorder of motor control, associated with a deficiency of dopamine in the nigrostriatal pathway.
In treating CNS disorders, it is often desired that a certain receptor type be activated or inhibited only in one part of the brain but the problem is that drugs are rarely brain region selective and will affect all of a receptor type throughout the brain. For example, many antipsychotic drugs (see Ch. 45) are D2 receptor antagonists, exerting a beneficial effect by blocking D2 receptors in the mesolimbic pathway. However, their D2 antagonist property also gives rise to their major side effect, which is to cause movement disorders, by simultaneously blocking D2 receptors in the nigrostriatal pathway.
Transgenic mice lacking D2 receptors show greatly reduced spontaneous movement, resembling Parkinson’s disease.
Behavioural effects
Administration of amphetamine to rats, which releases both dopamine and noradrenaline, causes a cessation of normal ‘ratty’ behaviour (exploration and grooming), and the appearance of repeated ‘stereotyped’ behaviour (rearing, gnawing and so on) unrelated to external stimuli. These amphetamine-induced motor disturbances in rats probably reflect hyperactivity in the nigrostriatal dopaminergic system, and are prevented by dopamine antagonists and by destruction of dopamine-containing cell bodies in the midbrain, but not by drugs that inhibit the noradrenergic system.
Amphetamine and cocaine (which act by inhibiting the dopamine transporter) and also other drugs of abuse (Ch. 48) activate mesolimbic dopaminergic ‘reward’ pathways to produce feelings of euphoria in humans. The main receptor involved appears to be D1, and transgenic mice lacking D1 receptors behave as though generally demotivated, with reduced food intake and insensitivity to amphetamine and cocaine (see Sibley, 1999).
Neuroendocrine function
The tuberohypophyseal dopaminergic pathway (see Fig. 38.3) is involved in the control of prolactin secretion. The hypothalamus secretes various mediators (mostly small peptides; see Ch. 32), which control the secretion of different hormones from the pituitary gland. One of these mediators, which has an inhibitory effect on prolactin release, is dopamine. This system is of clinical importance. Many antipsychotic drugs (see Ch. 45), by blocking D2 receptors, increase prolactin secretion and can cause breast development and lactation, even in males. Bromocriptine, a dopamine receptor agonist derived from ergot, is used clinically to suppress prolactin secretion by tumours of the pituitary gland.
Growth hormone production is increased in normal subjects by dopamine, but bromocriptine paradoxically inhibits the excessive secretion responsible for acromegaly (probably because it desensitises dopamine receptors, in contrast to the physiological release of dopamine, which is pulsatile) and has a useful therapeutic effect, provided it is given before excessive growth has taken place. It is now rarely used, as other agents are more effective (see Ch. 32). Bromocriptine and other dopamine agonists, such as cabergoline, enhance libido and sexual performance.
Vomiting
Pharmacological evidence strongly suggests that dopaminergic neurons have a role in the production of nausea and vomiting. Thus nearly all dopamine receptor agonists (e.g. bromocriptine) and other drugs that increase dopamine release in the brain (e.g. levodopa; Ch. 39) cause nausea and vomiting as side effects, while many dopamine antagonists (e.g. phenothiazines, metoclopramide; Ch. 29) have antiemetic activity. D2 receptors occur in the area of the medulla (chemoreceptor trigger zone) associated with the initiation of vomiting (Ch. 29), and are assumed to mediate this effect.
Dopamine in the CNS
5-Hydroxytryptamine
The occurrence and functions of 5-HT (serotonin) in the periphery are described in Chapter 15. Interest in 5-HT as a possible CNS transmitter dates from 1953, when Gaddum found that lysergic acid diethylamide (LSD), a drug known to be a powerful hallucinogen (see Ch. 47), acted as a 5-HT antagonist on peripheral tissues, and suggested that its central effects might also be related to this action. The presence of 5-HT in the brain was demonstrated a few years later. Even though brain accounts for only about 1% of the total body content, 5-HT is an important CNS transmitter (see Iversen et al., 2009; Muller & Jacobs, 2009). 5-HT is involved in various physiological processes including sleep, appetite, thermoregulation and pain perception as well as in disorders such as migraine, depression, anxiety, obsessive compulsive disorders, schizophrenia and drug abuse.
In its formation, storage and release, 5-HT resembles noradrenaline. Its precursor is tryptophan, an amino acid derived from dietary protein, the plasma content of which varies considerably according to food intake and time of day. Tryptophan is actively taken up into neurons, converted by tryptophan hydroxylase to 5-hydroxytryptophan (see Fig. 15.1), and then decarboxylated by a non-specific amino acid decarboxylase to 5-HT. Tryptophan hydroxylase can be selectively and irreversibly inhibited by p-chlorophenylalanine (PCPA). Availability of tryptophan and the activity of tryptophan hydroxylase are thought to be the main factors that regulate 5-HT synthesis. The decarboxylase is very similar, if not identical, to dopa decarboxylase, and does not play any role in regulating 5-HT synthesis. Following release, 5-HT is largely recovered by neuronal uptake, through a specific transporter (see Ch. 3) similar to, but not identical with, those that take up noradrenaline and dopamine. 5-HT reuptake is specifically inhibited by selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine and by many of the drugs that inhibit catecholamine uptake (e.g. tricyclic antidepressants). SSRIs (see Ch. 46) constitute an important group of antidepressant drugs. 5-HT is degraded almost entirely by monoamine oxidase (Fig. 15.1), which converts it to 5-hydroxyindole acetaldehyde, most of which is then dehydrogenated to form 5-hydroxyindole acetic acid (5-HIAA) and excreted in the urine.
5-HT Pathways in the CNS
The distribution of 5-HT-containing neurons (Fig. 38.6) resembles that of noradrenergic neurons. The cell bodies are grouped in the pons and upper medulla, close to the midline (raphe), and are often referred to as raphe nuclei. The rostrally situated nuclei project, via the medial forebrain bundle, to many parts of the cortex, hippocampus, basal ganglia, limbic system and hypothalamus. The caudally situated cells project to the cerebellum, medulla and spinal cord.
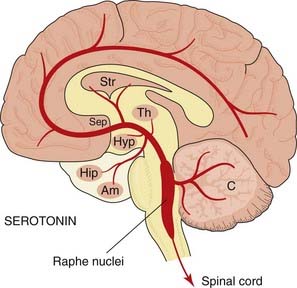
Fig. 38.6 Simplified diagram of the 5-hydroxytryptamine pathways in the brain, drawn as in Figure 38.1.
Abbreviations as in Figure 38.1.
5-HT Receptors in the CNS
The main 5-HT receptor types are shown in Table 15.1. All are G-protein-coupled receptors except for 5-HT3, which is a ligand-gated cation channel (see below). All are expressed in the CNS, and their functional roles have been extensively analysed. With some 14 identified subtypes plus numerous splice variants, and a large number of pharmacological tools of relatively low specificity, assigning clear-cut functions to 5-HT receptors is not simple. Detailed accounts of our present state of knowledge are given by Barnes & Sharp (1999) and Bockaert et al. (2006). Knowledge about the newer members of the family (5-HT5-7 receptors) is summarised in reviews by Woolley et al. (2004) and Hedlund & Sutcliffe (2004).
Certain generalisations can be made:
Functional Aspects
The precise localisation of 5-HT neurons in the brain stem has allowed their electrical activity to be studied in detail and correlated with behavioural and other effects produced by drugs thought to affect 5-HT-mediated transmission. 5-HT cells show an unusual, highly regular, slow discharge pattern, and are strongly inhibited by 5-HT1 receptor agonists, suggesting a local inhibitory feedback mechanism.
In vertebrates, certain physiological and behavioural functions relate particularly to 5-HT pathways (see Barnes & Sharp, 1999), namely:
Hallucinatory effects
Many hallucinogenic drugs (e.g. LSD; Ch. 47) are agonists at 5-HT2A receptors. It is suggested that a loss of cortical inhibition underlies the hallucinogenic effect, as well as certain behavioural effects in experimental animals, such as the ‘wet dog shakes’ that occur in rats when the 5-HT precursor 5-hydroxytryptophan is administered. Many antipsychotic drugs (Ch. 45) are antagonists at 5-HT2A receptors in addition to blocking dopamine D2 receptors. The psychostimulant properties of MDMA (‘ecstasy’; see Ch. 47) are due partly to its ability to release 5-HT. MDMA is taken up by the serotonin transporter, causing it to displace 5-HT from storage vesicles—a mechanism analogous to the action of amphetamine on noradrenergic nerve terminals (Ch. 14).
Sleep, wakefulness and mood
Lesions of the raphe nuclei, or depletion of 5-HT by PCPA administration, abolish sleep in experimental animals, whereas microinjection of 5-HT at specific points in the brain stem induces sleep. 5-HT7 receptor antagonists inhibit ‘rapid-eye-movement’ (REM) sleep and increase the latency to onset of REM sleep. Attempts to cure insomnia in humans by giving 5-HT precursors (tryptophan or 5-hydroxytryptophan) have, however, proved unsuccessful. There is strong evidence that 5-HT, as well as noradrenaline, may be involved in the control of mood (see Ch. 46), and the use of tryptophan to enhance 5-HT synthesis has been tried in depression, with equivocal results.
Feeding and appetite
In experimental animals, 5-HT1A agonists such as 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) cause hyperphagia, leading to obesity. Antagonists acting on 5-HT2 receptors, including several antipsychotic drugs used clinically, also increase appetite and cause weight gain. On the other hand, antidepressant drugs that inhibit 5-HT uptake (see Ch. 46) cause loss of appetite.
Sensory transmission
After lesions of the raphe nuclei or administration of PCPA, animals show exaggerated responses to many forms of sensory stimulus. They are startled much more easily, and also quickly develop avoidance responses to stimuli that would not normally bother them. It appears that the normal ability to disregard irrelevant forms of sensory input requires intact 5-HT pathways. The ‘sensory enhancement’ produced by hallucinogenic drugs may be partly due to loss of this gatekeeper function of 5-HT. 5-HT also exerts an inhibitory effect on transmission in the pain pathway, both in the spinal cord and in the brain, and there is a synergistic effect between 5-HT and analgesics such as morphine (see Ch. 41). Thus, depletion of 5-HT by PCPA, or selective lesions to the descending 5-HT-containing neurons that run to the dorsal horn, antagonise the analgesic effect of morphine, while inhibitors of 5-HT uptake have the opposite effect.
Other possible roles
Other putative roles of 5-HT include various autonomic and endocrine functions, such as the regulation of body temperature, blood pressure and sexual function. Further information can be found in Azmitia & Whitaker-Azmitia (1995) and Iversen et al. (2009).
Clinically Used Drugs
Several classes of drugs used clinically influence 5-HT-mediated transmission. They include:
5-Hydroxytryptamine in the CNS
Acetylcholine
There are numerous cholinergic neurons in the CNS, and the basic processes by which ACh is synthesised, stored and released are the same as in the periphery (see Ch. 13). Various biochemical markers have been used to locate cholinergic neurons in the brain, the most useful being choline acetyltransferase, the enzyme responsible for ACh synthesis, and the transporters that capture choline and package ACh, which can be labelled by immunofluorescence. Biochemical studies on ACh precursors and metabolites are generally more difficult than corresponding studies on other amine transmitters, because the relevant substances, choline and acetate, are involved in many processes other than ACh metabolism.
Cholinergic Pathways in the Cns
Acetylcholine is very widely distributed in the brain, occurring in all parts of the forebrain (including the cortex), midbrain and brain stem, although there is little in the cerebellum. Cholinergic neurons in the forebrain and brain stem send diffuse projections to many parts of the brain (see Fig. 38.7). Cholinergic neurons in the forebrain lie in a discrete area, forming the magnocellular forebrain nuclei (so called because the cell bodies are conspicuously large). Degeneration of one of these, the nucleus basalis of Meynert, which projects mainly to the cortex, is associated with Alzheimer’s disease (Ch. 39). Another cluster, the septohippocampal nucleus, provides the main cholinergic input to the hippocampus, and is involved in memory. In addition, there are—in contrast to the monoamine pathways—many local cholinergic interneurons, particularly in the corpus striatum, these being important in relation to Parkinson’s disease and Huntington’s chorea (Ch. 39).
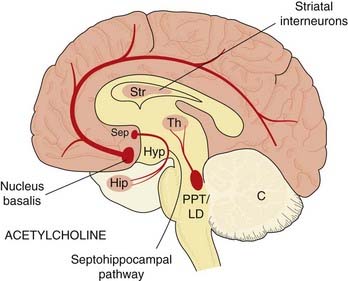
Fig. 38.7 Simplified diagram of the acetylcholine pathways in the brain, drawn as in Figure 38.1.
PPT/LD, pedunculopontine and laterodorsal tegmental nuclei; other abbreviations as in Figure 38.1.
Acetylcholine Receptors
Acetylcholine acts on both muscarinic (G-protein-coupled) and nicotinic (ionotropic) receptors in the CNS (see Ch. 13).
The muscarinic ACh receptors (mAChRs) in the brain are predominantly of the Gq-coupled M1 class (i.e. M1, M3 and M5 subtypes; see Ch. 13). Activation of these receptors can result in excitation through blockade of M-type (KCNQ/Kv7) K+ channels (see Delmas & Brown, 2005). Gi/Go-coupled M2 and M4 receptors, on the other hand, are inhibitory through activation of inwardly rectifying K+ channels and inhibition of voltage-sensitive Ca2+ channels. mAChRs on cholinergic terminals function to inhibit ACh release, and muscarinic antagonists, by blocking this inhibition, markedly increase ACh release. Many of the behavioural effects associated with cholinergic pathways seem to be produced by ACh acting on mAChRs.
Nicotinic ACh receptors (nAChRs) are ligand-gated cation channels permeable to Na+, K+ and Ca2+ ions (see Ch. 13). They are pentamers and can be formed as homomeric or heteromeric combinations of α (α2–7) and β (β2–4) subunits (Ch. 3; see Gotti et al., 2008) distributed widely throughout the brain (see Table 38.2). The heteromeric α4β2 and the homomeric α7 subtypes are the most extensively characterised. The lack of subtype-specific ligands and the fact that some neurons express multiple subtypes has made the elucidation of the functions of each receptor subtype extremely difficult. Nicotine (see Ch. 48) exerts its central effects by agonist action on nAChRs.
Table 38.2 Presence of nicotinic receptors of different subunit composition in selected regions of the central nervous system
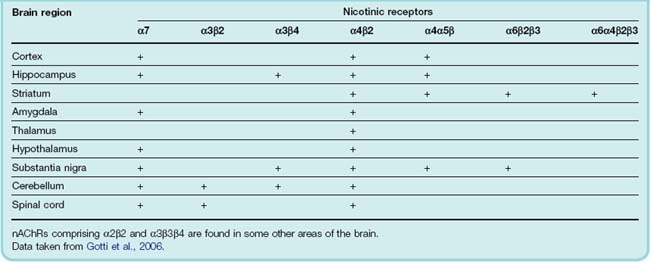
For the most part, nAChRs are located presynaptically and act usually to facilitate the release of other transmitters such as glutamate, dopamine and GABA.2 In a few situations, they function postsynaptically to mediate fast excitatory transmission, as in the periphery.
Many of the drugs that block nAChRs (e.g. tubocurarine; see Ch. 13) do not cross the blood–brain barrier, and even those that do (e.g. mecamylamine) produce only modest CNS effects. Various nAChR knockout mouse strains have been produced and studied. Deletion of the various CNS-specific nAChR subtypes generally has rather little effect, although some cognitive impairment can be detected. Mutations in nAChRs may be the cause of some forms of epilepsy and changes in nAChR expression may occur in disorders such as schizophrenia, attention deficit hyperactivity disorder, depression and anxiety, as well as following neurodegeneration in Alzheimer’s and Parkinson’s diseases.
Functional Aspects
The functional roles of cholinergic pathways have been deduced mainly from studies of the action of drugs that mimic, accentuate or block the actions of ACh, and from studies of transgenic animals in which particular AChRs were deleted or mutated (see Cordero-Erausquin et al., 2000; Hogg et al., 2003).
The main functions ascribed to cholinergic pathways are related to arousal, learning and memory, and motor control. The cholinergic projection from the ventral forebrain to the cortex is thought to mediate arousal, whereas the septohippocampal pathway is involved in learning and short-term memory (see Hasselmo, 2006). Cholinergic interneurons in the striatum are involved in motor control (see Ch. 39).
Muscarinic agonists have been shown to restore partially learning and memory deficits induced in experimental animals by lesions of the septohippocampal cholinergic pathway. Hyoscine, a muscarinic antagonist, impairs memory in human subjects and causes amnesia when used as preanaesthetic medication. M1 receptor knockout mice, however, show only slight impairment of learning and memory (see Wess, 2004).
Nicotine increases alertness and also enhances learning and memory, as do various synthetic agonists at neuronal nAChRs. Conversely, CNS-active nAChR antagonists such as mecamylamine cause detectable, although slight, impairment of learning and memory. Transgenic mice with disruption of brain nAChRs are only slightly impaired in spatial learning tasks.
In conclusion, both nAChRs and mAChRs may play a role in learning and memory, while nAChRs also mediate behavioural arousal. Receptor knockout mice are surprisingly little affected, suggesting that alternative mechanisms may be able to compensate for the loss of ACh receptor signalling.
The importance of cholinergic neurons in neurodegenerative conditions such as dementia and Parkinson’s disease is discussed in Chapter 39. The role of nAChRs in modulating pain transmission in the CNS is described in Chapter 41.
Acetylcholine in the CNS
Purines
Both adenosine and ATP act as transmitters and/or modulators in the CNS (for review, see Fredholm et al., 2005; Khahk & North, 2006) as they do in the periphery (Ch. 16). Mapping the pathways is difficult, because purinergic neurons are not easily identifiable histochemically, but it is likely that adenosine serves as a very widespread neuromodulator, while ATP has more specific synaptic functions as a fast transmitter and as a local modulator.
Adenosine is produced intracellularly from ATP. It is not packaged into vesicles but is released mainly by carrier-mediated transport. Because the intracellular concentration of ATP (several mmol/l) greatly exceeds that of adenosine, conversion of a small proportion of ATP results in a large increase in adenosine. ATP is packaged into vesicles and released by exocytosis as a conventional transmitter, but can also leak out of cells in large amounts under conditions of tissue damage. In high concentrations, ATP can act as an excitotoxin (like glutamate; see Ch. 39) and cause further neuronal damage. It is also quickly converted to adenosine, which exerts a protective effect. These special characteristics of adenosine metabolism suggest that it serves mainly as a safety mechanism, protecting the neurons from damage when their viability is threatened, for example by ischaemia or seizure activity.
Adenosine produces its effects through G-protein-coupled adenosine A receptors (see Ch. 16). For ATP there are two forms of receptor—P2X and P2Y receptors (see Ch. 16 also). P2X receptors are trimeric ligand-gated cation channels that can be homomeric or heteromeric in composition whereas P2Y receptors are G-protein coupled.
There are four adenosine receptors—A1, A2A, A2B and A3—distributed throughout the CNS. The overall effect of adenosine, or of various adenosine receptor agonists, is inhibitory, leading to effects such as drowsiness and sedation, motor incoordination, analgesia and anticonvulsant activity. Xanthines, such as caffeine (Ch. 47), which are antagonists at A2 receptors, produce arousal and alertness.
While there is little doubt that purinergic signalling plays a major role in CNS function, our understanding is still very limited. There is optimism that purinergic receptor ligands—both agonists and antagonists—will prove useful in a wide range of CNS disorders (see Burnstock, 2008).
Histamine
Histamine is present in the brain in much smaller amounts than in other tissues, such as skin and lung, but undoubtedly serves a neurotransmitter role (see Brown et al., 2001). The cell bodies of histaminergic neurons, which also synthesise and release a variety of other transmitters, are restricted to a small part of the hypothalamus, and their axons run to virtually all parts of the brain. Unusually, no uptake mechanism for histamine is present, its action being terminated instead by enzymic methylation.
Histamine acts on at least three types of receptor (H1–3; Ch. 17) in the brain (the evidence for H4 receptors in brain is still rather flimsy). They occur in most brain regions and are all G-protein coupled—H1 receptors to Gq, H2 to Gs and H3 to Gi/Go. H3 receptors are inhibitory autoreceptors on histamine-releasing neurons.
Like other monoamine transmitters, histamine is involved in many different CNS functions. Histamine release follows a distinct circadian pattern, the neurons being active by day and silent by night. H1 receptors in the cortex and reticular activating system contribute to arousal and wakefulness, and H1 receptor antagonists produce sedation (see Ch. 43). Other functions ascribed to histamine include control of food and water intake, and thermoregulation, but these are less well characterised. Antihistamines are widely used to control nausea and vomiting, for example in motion sickness and middle ear disorders, as well as to induce sleep.
Other CNS Mediators
We now move from the familiar neuropharmacological territory of the ‘classic’ monoamines to some of the frontier towns, bordering on the Wild West. Useful drugs are still few and far between in this area, and if applied pharmacology is your main concern, you can safely skip the next part and wait a few years for law and order to be established.
Melatonin
Melatonin (N-acetyl-5-methoxytryptamine) (reviewed by Dubocovich et al., 2003) is synthesised exclusively in the pineal, an endocrine gland that plays a role in establishing circadian rhythms. The gland contains two enzymes, not found elsewhere, which convert 5-HT by acetylation and O-methylation to melatonin, its hormonal product.
There are two well-defined melatonin receptors (MT1 and MT2) which are G-protein-coupled receptors—both coupling to Gi/Go—found mainly in the brain and retina but also in peripheral tissues (see Jockers et al., 2008). Another type (termed MT3) has been suggested to be the enzyme quinone reductase 2 (QR2). The function of the interaction between melatonin and QR2 is still unclear.
Melatonin secretion (in all animals, whether diurnal or nocturnal in their habits) is high at night and low by day. This rhythm is controlled by input from the retina via a noradrenergic retinohypothalamic tract that terminates in the suprachiasmatic nucleus (SCN) in the hypothalamus, a structure often termed the ‘biological clock’, which generates the circadian rhythm. Activation of MT1 receptors inhibits neuronal firing in the SCN and prolactin secretion from the pituitary. Activation of MT2 receptors phase shifts circadian rhythms generated within the SCN.
Given orally, melatonin is well absorbed but quickly metabolised, its plasma half-life being a few minutes. It has been promoted as a means of controlling jet lag, or of improving the performance of night-shift workers, based on its ability to reset the circadian clock. A single dose appears to have the effect of resynchronising the physiological secretory cycle, although it is not clear how this occurs. Ramelteon, an agonist at MT1 and MT2 receptors, is used to treat insomnia (see Ch. 43) and agomelatine, which has agonist actions at MT1 and MT2 receptors as well as antagonist actions at 5-HT2c receptors, is a novel antidepressant drug (see Ch. 46)
Nitric Oxide
Nitric oxide (NO) as a peripheral mediator is discussed in Chapter 20. Its significance as an important chemical mediator in the nervous system has demanded a considerable readjustment of our views about neurotransmission and neuromodulation (for review, see Garthwaite, 2008). The main defining criteria for transmitter substances—namely that neurons should possess machinery for synthesising and storing the substance, that it should be released from neurons by exocytosis, that it should interact with specific membrane receptors and that there should be mechanisms for its inactivation—do not apply to NO. Moreover, it is an inorganic gas, not at all like the kind of molecule we are used to. The mediator function of NO is now well established (Zhou & Zhu, 2009). NO diffuses rapidly through cell membranes, and its action is not highly localised. Its half-life depends greatly on the chemical environment, ranging from seconds in blood to several minutes in normal tissues. The rate of inactivation of NO (see Ch. 20, reaction 20.1) increases disproportionately with NO concentration, so low levels of NO are relatively stable. The presence of superoxide, with which NO reacts (see below), shortens its half-life considerably.
Nitric oxide in the nervous system is produced mainly by the constitutive neuronal form of nitric oxide synthase (nNOS; see Ch. 20), which can be detected either histochemically or by immunolabelling. This enzyme is present in roughly 2% of neurons, both short interneurons and long-tract neurons, in virtually all brain areas, with particular concentrations in the cerebellum and hippocampus. It occurs in cell bodies and dendrites, as well as in axon terminals, suggesting that NO may be produced both pre- and postsynaptically. nNOS is calmodulin dependent and is activated by a rise in intracellular Ca2+ concentration, which can occur by many mechanisms, including action potential conduction and neurotransmitter action, especially by glutamate activation of Ca2+-permeable NMDA receptors. NO is not stored, but released as it is made. Many studies have shown that NO production is increased by activation of synaptic pathways, or by other events, such as brain ischaemia (see Ch. 39).
Nitric oxide exerts pre- and postsynaptic actions on neurons as well as acting on glial cells (Garthwaite, 2008). It produces its effects in two main ways:
There is good evidence that NO plays a role in synaptic plasticity (see Ch. 37), because long-term potentiation and depression are reduced or prevented by NOS inhibitors and are absent in transgenic mice in which the nNOS gene has been disrupted.
Based on the same kind of evidence, NO is also believed to play an important part in the mechanisms by which ischaemia causes neuronal death (see Ch. 39). There is also evidence that it may be involved in other processes, including neurodegeneration in Parkinson’s disease, senile dementia and amyotrophic lateral sclerosis, and the local control of blood flow linked to neuronal activity.
Carbon monoxide (CO) is best known as a poisonous gas present in vehicle exhaust, which binds strongly to haemoglobin, causing tissue anoxia. However, it is also formed endogenously and has many features in common with NO (see Barañano et al., 2001). Neurons and other cells contain a CO-generating enzyme, haem oxygenase, and CO, like NO, activates guanylyl cyclase.
The role of CO as a CNS mediator is not well established, but there is some evidence that it plays a role in memory mechanisms in the hippocampus (see Cutajar & Edwards, 2007).
Lipid Mediators
The formation of arachidonic acid, and its conversion to eicosanoids (mainly prostaglandins, leukotrienes and hydroxyeicosatetraenoic acids (HETEs); see Ch. 17) and to endocannabinoids, anandamide and 2-arachidonoylglycerol (see Ch. 18), also take place in the CNS (for reviews, see Piomelli, 1995; Pertwee, 2008).
Phospholipid cleavage, leading to arachidonic acid production, occurs in neurons in response to receptor activation by many different mediators, including neurotransmitters. The arachidonic acid so formed can act directly as an intracellular messenger, controlling both ion channels and various parts of the protein kinase cascade (see Ch. 3), producing both rapid and delayed effects on neuronal function. Both arachidonic acid itself and its products escape readily from the cell of origin and can affect neighbouring structures, including presynaptic terminals (retrograde signalling) and adjacent cells (paracrine signalling), by acting on receptors or by acting directly as intracellular messengers. Figure 38.8 shows a schematic view of the variety of different roles these agents can play at the synapse.
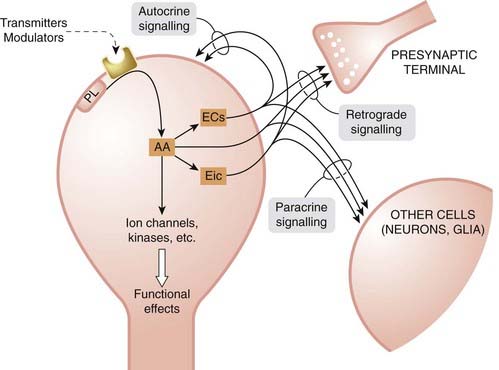
Fig. 38.8 Postulated modes of signalling by lipid mediators.
Arachidonic acid (AA) is formed by receptor-mediated cleavage of membrane phospholipid. It can act directly as an intracellular messenger on ion channels or components of different kinase cascades, producing various long- and short-term effects. It can also be converted to eicosanoids (prostaglandins, leukotrienes or hydroxyeicosatetraenoic acids [HETEs]) or to the endocannabinoids (ECs), anandamide and 2-arachidonoylglycerol. HETEs can also act directly as intracellular messengers. All these mediators diffuse out of the cell, and exert effects on presynaptic terminals and neighbouring cells, acting either on extracellular receptors or intracellularly. There are examples of most of these modes of signalling but only limited information about their functional significance in the nervous system. Eic, eicosanoids; PL, membrane phospholipid.
Arachidonic acid can be metabolised to eicosanoids, some of which (principally the HETEs) can also act as intracellular messengers acting in the same cell. Eicosanoids can also exert an autocrine effect via membrane receptors expressed by the cell (see Ch. 17). The eicosanoids play important roles in neural function including pain, temperature regulation, sleep induction, synaptic plasticity and spatial learning.
It is now generally accepted that the endocannabinoids act as retrograde synaptic messengers. They are synthesised and secreted in response to a rise in intracellular Ca2+ and activate presynaptic CB1 receptors resulting in an inhibition of the release of neurotransmitters such as glutamate and GABA (see Vaughan & Christie, 2005). CB1 receptors are widely distributed in the brain and spinal cord whereas CB2 receptor expression is much less. Agonists at CB1 receptors have therapeutic potential for the treatment of vomiting, pain (CB2 receptor agonists may also be effective in some pain states), muscle spasms as occur in conditions such as multiple sclerosis and anxiety, as well as in other brain disorders including Alzheimer’s disease and tardive dyskinesias (see Pertwee, 2008). The CB1 receptor antagonist, rimonabant, was introduced as an antiobesity agent but subsequently had to be withdrawn because of negative effects on mood (see Ch. 18). One surprise in this field has been the discovery that anandamide, besides being an agonist at cannabinoid receptors, also activates TRPV1 channels (see Ch. 41) which are involved in the response of peripheral sensory nerve terminals to painful stimuli.
Other transmitters and modulators
Purines
Histamine
Melatonin
Nitric oxide (see Ch. 20)
Lipid mediators
A Final Message
In the last two chapters, we have taken a long and tortuous tour through the brain and its chemistry, with two questions at the back of our minds. What mediators and what receptors play a key role in what brain functions? How does the information relate to existing and future drugs that aim to correct malfunctions? Through the efforts of a huge army of researchers deploying an arsenal of powerful new techniques, the answers to these questions are slowly being produced. The array of potential CNS targets—comprising multiple receptor subtypes, many with the added complexity of heteromeric assemblies, splice variants, etc., along with regulatory mechanisms that control their expression and localisation—continues to grow in complexity. Speculation about the best target to aim at in order to ameliorate the effect of a particular brain malfunction, such as stroke or schizophrenia, has become less focused, even if better informed, than it was two decades ago. In the ensuing chapters in this section, we shall find that most of the therapeutic successes have come from chance discoveries that were followed up empirically; few have followed a logical, mechanism-based route to success. The optimistic view is that this is changing, and that future therapeutic discoveries will depend less on luck and more on molecular logic. But the revolution is slow in coming. One of the key problems, perhaps, is that the brain puts cells, organelles and molecules exactly where they are needed, and uses the same molecules to perform different functions in different locations. Drug discovery scientists are getting quite good at devising molecule-specific ligands (see Ch. 60), but we lack delivery systems able to target them anatomically even to macroscopic brain regions, let alone to specific cells and subcellular structures.
References and Further Reading
Davis K.L., Charney D., Coyle J.T., Nemeroff C., editors. Neuropsychopharmacology: the fifth generation of progress. Philadelphia: Lippincott, Williams & Wilkins, 2002. (A 2000-page monster with excellent and authoritative articles on basic and clinical aspects)
Iversen L.L., Iversen S.D., Bloom F.E., Roth R.H. Introduction to neuropsychopharmacology. New York: Oxford University Press; 2009. (Clear and well-written textbook giving more detailed information on many topics covered in this chapter)
Nestler E.J., Hyman S.E., Malenka R.C. Molecular neuropharmacology: a foundation for clinical neuroscience, second ed. New York: McGraw-Hill; 2008. . (Good modern textbook)
Bylund D.B. Receptors for norepinephrine and signal transduction pathways. In: Ordway G.A., Schwartz M.A., Frazer A., editors. Brain norepinephrine. London: Cambridge University Press, 2007.
Head G.A., Mayorov D.N. Imidazoline receptors, novel agents and therapeutic potential. Cardiovasc. Hematol. Agents Med. Chem.. 2006;4:17-32. Provides an update on the elusive imidazoline receptors)
Björklund A., Dunnett S.B. Dopamine neuron systems in the brain: an update. Trends Neurosci.. 2007;30:194-202. (Short review of the anatomy of dopaminergic neurons in the central nervous system)
De Mei C., Ramos M., Iitaka C., Borrelli E. Getting specialized: presynaptic and postsynaptic dopamine D2 receptors. Curr. Opin. Pharmacol.. 2009;9:53-58.
Girault J.-A., Greengard P. The neurobiology of dopamine signalling. Arch. Neurol.. 2004;61:641-644. (Short review article)
Missale C., Nash S.R., Robinson S.W., et al. Dopamine receptors: from structure to function. Physiol. Rev.. 1998;78:198-225. (Comprehensive review article)
Sibley D.R. New insights into dopaminergic receptor function using antisense and genetically altered animals. Annu. Rev. Pharmacol. Toxicol.. 1999;39:313-341.
Tarazi F.I., Zhang K., Baldessarini R.J. Dopamine D4 receptors: beyond schizophrenia J. Recept. Signal Transduct. 24:2004:131-147 (Dismisses link between D4 receptor polymorphism and schizophrenia, suggesting possible link with attention deficit hyperactivity disorder, impulsivity and cognitive function)
Thapar A., Langley K., Owen M.J., O’Donovan M.C. Advances in genetic findings on attention deficit hyperactivity disorder 37. 2007 Psychol. Med.
Azmitia E.C., Whitaker-Azmitia P.M. Anatomy, cell biology and plasticity of the serotonergic system. In: Bloom F.E., Kupfer D.J., editors. Psychopharmacology: a fourth generation of progress. New York: Raven Press, 1995. (General review article)
Barnes N.M., Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083-1152. (Detailed compilation of data relating to distribution, pharmacology and function of 5-HT receptors in the CNS; useful information source but not particularly illuminating)
Bockaert J., Claeysen S., Becamel C., et al. Neuronal 5-HT metabotropic receptors: fine-tuning of their structure, signaling, and roles in synaptic modulation. Cell Tissue Res.. 2006;326:553-572.
Hedlund P.B., Sutcliffe J.G. Functional, molecular and pharmacological advances in 5-HT7 receptor research Trends Pharmacol. Sci. 25:2004:481-486 (Reviews current understanding of role of 5-HT7 receptors, including data from receptor knockout mice)
Jensen A.A., Davies P.A., Bräuner-Osborne H., Krzywkowski K. 3B but which 3B? And that’s just one of the questions: the heterogeneity of human 5-HT3 receptors Trends Pharmacol. Sci. 29:2008:437-444 (Discusses the potential complexity of 5-HT3 receptors now that new subunits have been discovered)
Muller C., Jacobs B. Handbook of behavioral neurobiology of serotonin vol. 18. 2009 Academic Press Oxford. (Extensive coverage of the role of 5-HT in the brain) (Handbook of behavioral neuroscience)
Peters J.A., Hales T.G., Lambert J.J. Molecular determinants of single-channel conductance and ion selectivity in the Cys-loop family: insights from the 5-HT3 receptor. Trends Pharmacol. Sci.. 2005;26:587-594. (For those who thought ligand-gated ion channels were just simple pores opened by neurotransmitters, this review will contain a few surprises!)
Woolley M.L., Marsden C.A., Fone K.C. 5-HT6 receptors Curr. Drug Targets CNS Neurol. Disord. 3:2004:59-79 (General review article focusing on the many possible clinical applications of 5-HT6 receptor agonists and antagonists)
Cordero-Erausquin M., Marubio L.M., Klink R., Changeux J.-P. Nicotinic receptor function: new perspectives from knockout mice. Trends Pharmacol. Sci.. 2000;21:211-217. (Short review article)
Delmas P., Brown D.A. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci.. 2005;6:850-862. (Gives information on the functional significance of the ‘M-current’ and the therapeutic potential of drugs that modify it)
Gotti C., Zoli M., Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol. Sci.. 2008;27:482-491.
Hasselmo M.E. The role of acetylcholine in learning and memory. Curr. Opin. Neurobiol.. 2006;16:710-715.
Hogg R.C., Raggenbass M., Bertrand D. Nicoticin acetylcholine receptors: from structure to brain function. Rev. Physiol. Biochem. Pharmacol.. 2003;147:1-46. (General review of molecular and functional properties of brain nAChRs)
Khahk B.S., Henderson G. Modulation of fast synaptic transmission by presynaptic ligand-gated cation channels. J. Auton. Nerv. Syst.. 2000;81:110-121. (Describes how activation of presynaptic ligand-gated cation channels can either enhance or inhibit neurotransmitter release)
Wess J. Muscarinic acetylcholine receptor knockout mice: novel phenotypes and clinical implications. Annu. Rev. Pharmacol. Toxicol.. 2004;44:423-450. (Description of functional effects of deleting various peripheral and central mAChR isoforms)
Barañano D.E., Ferris C.D., Snyder S.H. Atypical neural messengers. Trends Neurosci.. 2001;24:99-106. (Short review on some established mediators such as NO, and some speculative ones)
Brown R.E., Stevens D.R., Haas H.L. The physiology of brain histamine. Prog. Neurobiol.. 2001;63:637-672. (Useful review article)
Burnstock G. Purinergic signalling and disorders of the central nervous system. Nat. Rev. Drug Discov.. 2008;7:575-590. (Extensive discussion of the therapeutic potential of drugs acting at purinergic receptors)
Cutajar M.C., Edwards T.M. Evidence for the role of endogenous carbon monoxide in memory processing. J. Cogn. Neurosci.. 2007;19:557-562.
Dubocovich M.L., Rivera-Bermudez M.A., Gerdin M.J., Masana M.I. Molecular pharmacology, regulation and function of mammalian melatonin receptors. Front. Biosci.. 2003;8:1093-1108.
Fredholm B.B., Chen J.F., Masino S.A., Vaugeois J.M. Actions of adenosine at its receptors in the CNS: insights from knockouts and from drugs. Annu. Rev. Pharmacol. Toxicol.. 2005;45:385-412.
Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur. J. Neurosci.. 2008;27:2783-2802.
Jockers R., Maurice P., Boutin J.A., Delagrange P. Melatonin receptors, heterodimerization, signal transduction and binding sites: what’s new? Br. J. Pharmacol.. 2008;154:1182-1195.
Khahk B.S., North R.A. P2X receptors as cell-surface ATP sensors in health and disease. Nature. 2006;442:527-532.
Pertwee R.G. Ligands that target cannabinoid receptors in the brain: from THC to anandamide and beyond. Addic. Biol.. 2008;13:147-159.
Piomelli D. Arachidonic acid. In: Bloom F.E., Kupfer D.J., editors. Psychopharmacology: a fourth generation of progress. New York: Raven Press, 1995. (Excellent review article)
Vaughan C.W., Christie M.J. Retrograde signalling by endocannabinoids. Handb. Exp. Pharmacol.. 2005;168:367-383.
Zhou L., Zhu D.-Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation and clinical implications. Nitric Oxide. 2009;20:223-230.
1They are, if you like, voices from the nether regions, which make you happy or sad, sleepy or alert, cautious or adventurous, energetic or lazy, although you do not quite know why—very much the stuff of mental illness.
2See Khahk & Henderson, 2000, for a description of how presynaptic cation-selective ligand-gated channels can, under different circumstances, facilitate or enhance neurotransmitter release.