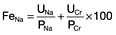Kidney involvement in other diseases
Polyarteritis nodosa (PAN)
Classical PAN is a multisystem disorder (see also p. 543). Aneurysmal dilatation of medium-sized arteries may be seen on renal arteriography. The condition is more common in men and in the elderly and, typically, the patient is ANCA negative. Hypertension, polyneuropathy and features indicating ischaemic infarction of various organs including the kidney are presenting features. It may be associated with drug use and hepatitis B infection. This form of polyangiitis is associated with slowly progressive CKD, often accompanied by severe hypertension. Rapidly progressive kidney failure is rare. Treatment with immunosuppression is less effective than it is for microscopic polyangiitis.
Systemic sclerosis (scleroderma)
Systemic sclerosis (scleroderma) is a chronic, multisystem disease characterized by fibrosis and vasculopathy of the skin and visceral organs. Of the patients, 10% develop scleroderma renal crisis, which is characterized by accelerated hypertension, rapidly progressive kidney failure and proteinuria.
Histopathological changes occur in the arcuate and interlobular arteries. Characteristically in the acute stage there are fibrin thrombi and areas of fibrinoid necrosis; these are followed by ‘onion skin’ hypertrophy of the arteries in the chronic stage. The treatment of choice is ACE inhibitors, which have led to remarkable improvement of outcomes in scleroderma renal crisis. Death is now rare, and <50% of patients progress to ESKD.
Haemolytic uraemic syndrome (HUS)
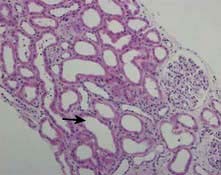
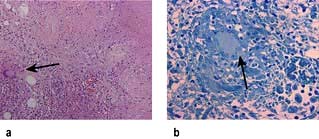
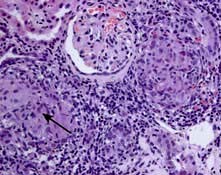
HUS is characterized by intravascular haemolysis with red-cell fragmentation (microangiopathic haemolysis), thrombocytopenia and acute kidney injury due to thrombosis in small arteries and arterioles (Fig. 12.25). These features are also seen in disseminated intravascular coagulation, but coagulation tests are typically normal in HUS.
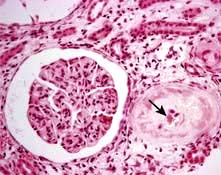
Figure 12.25 Typical haemolytic uraemic syndrome (HUS) renal lesion – light microscopy. Arrow shows microthrombi.
Diarrhoea-associated HUS (D+HUS)
This often follows a febrile illness, particularly gastroenteritis and usually associated with Escherichia coli, notably strain O157. This strain of E. coli produces verocytotoxin (shiga toxin), which has an A unit and five B units. The A unit is pathogenic by inhibiting protein synthesis and initiating endothelial damage. The role of B units is to facilitate the entry of the A unit into the endothelial cells by binding to a receptor (Gb3) on the endothelial cell. The toxins are transported to endothelial cells from the gut on neutrophils. Most patients with D+HUS recover renal function, but supportive care including maintenance of fluid and electrolyte balance, antihypertensive medication, nutritional support and dialysis is commonly required. Plasmapheresis is not beneficial but is usually tried as a last resort. About 5% die during the acute episode, 5% develop ESKD and 30% exhibit evidence of long-term damage with persistent proteinuria. Antibiotic and antimotility agents for the diarrhoea increase the risk of HUS and its complications.
Recently, an outbreak caused by shiga toxin-producing Escherichia coli O104:H4 (new strain) was reported in Germany and other European countries. In this outbreak, HUS cases were unusually high and associated with significant morbidity and mortality (see p. 121). Severe neurological complications were seen; immunoadsorption was successful in many cases.
Recurrent episodes of HUS have been described in the same individual, and familial forms of the disease (with both recessive and dominant inheritance) exist.
Non-diarrhoeal-induced form of HUS (D–HUS)
Also called atypical HUS (aHUS), this probably is a complement-driven illness due to a deficiency of complement factor H (CFH) or complement factor I (CFI). Factor H is a soluble protein produced by the liver, which regulates the activity of the alternative complement activation pathway; in particular, it protects host cell surfaces from complement-mediated damage. In some families with D–HUS, a mutation has been traced to another complement regulatory protein, CD46 (previously known as membrane cofactor protein, MCP). This protein is highly expressed in the kidney and normally prevents glomerular C3 activation. A loss of function mutation in CD46 results in unopposed complement activation and development of HUS. Functional deficiency of these factors can be acquired due to autoantibody formation either as an isolated phenomenon or as part of a rheumatic autoimmune disease such as SLE. A loss of function mutation in thrombomodulin (a membrane-bound anticoagulant glycoprotein) has been identified as an alternative complement pathway. Rarely gain of function mutations can affect genes encoding the alternative pathway C3 convertase components, CFB and C3. CFB mutations, which lead to chronic alternative-pathway activation, occur in only 1–2% of patients with D–HUS. About 4–10% of patients have heterozygous mutations in C3, usually with low C3 levels. Most mutations reduce C3b binding to CFH and CD46, which severely impairs degradation of mutant C3b.
Treatment
Treatment is often very difficult because of severe hypertension and the possibility of frequent recurrences. The course of the disease is often indolent and progressive. Plasmapheresis or plasma infusion is still the only therapy used in the majority of patients. C5 activation is one of the critical steps in the activation of complement cascade. Eculizumab, a monoclonal humanized anti-C5 antibody, has shown success in patients with D–HUS either dependent or refractory to plasmapheresis therapy. Liver transplantation is potentially the only curative treatment in patients harbouring CFH and CFI mutations.
Sporadic cases of D–HUS
These can be associated with pregnancy, SLE, scleroderma, malignant hypertension, metastatic cancer, HIV infection and various drugs including oral contraceptives, ciclosporin, tacrolimus, chemotherapeutic agents (e.g. cisplatin, mitomycin C, bleomycin) and heparin. Treatment is supportive with removal of the offending agent or specific treatment of the underlying cause. There is no evidence in favour of plasma infusion or plasmapheresis in these sporadic cases but it is tried, usually as a last resort.
Pneumococcus-associated HUS
This rare complication of Streptococcus pneumoniae infection was previously associated with a high morbidity and mortality. This organism produces an enzyme (possibly neuroaminidase) which can expose an antigen (Thomsen antigen) present on RBCs, platelets and glomeruli. Antibodies to the Thomsen antigen result in an antigen-antibody reaction and can lead to HUS and anaemia. The improved outcome is due to increasing awareness of this complication, judicious use of blood products (washed blood products) and avoiding plasma infusion or plasmapheresis.
Thrombotic thrombocytopenic purpura (TTP)
TTP (see p. 420) is characterized by microangiopathic haemolysis, renal failure and evidence of neurological disturbance. Young adults are most commonly affected.
Antiphospholipid syndrome (APS)
In antiphospholipid syndrome (APS, see p. 538), the binding of antiphospholipid antibodies (aPL) to beta 2 glycoprotein I (β2GPI) induces endothelial cell–leukocyte adhesion and thrombus formation by the inhibition of eNOS. The inhibition of eNOS is caused by antibody recognition and dimerization of β2GPI and impairment of eNOS phosphorylation.
The central feature of APS is recurrent thrombosis (both venous and arterial) and fetal loss in the presence of antiphospholipid antibodies. Such antibodies may be primary or secondary to infections (HIV, hepatitis C) or autoimmune disease (SLE). Some 50% have renal involvement with proteinuria. Thrombotic microangiopathy is a rare but well-recognized presentation. In some cases, a lupus nephritis-like (usually membranous GN) lesion is seen. The only proven treatment for APS is warfarin with an INR of 3–4. Use of steroids or plasmapheresis is reserved for patients with APS and life-threatening renal involvement with thrombotic microangiopathy. Treatment is variably successful (30–70%).
Multiple myeloma
Acute kidney injury (AKI) is relatively common in myeloma, occurring in 20–30% of affected individuals at the time of diagnosis, and is mainly due to the nephrotoxic effects of the abnormal immunoglobulins. It is often irreversible. The following types of renal lesions are associated with myeloma.
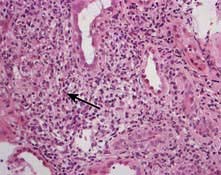
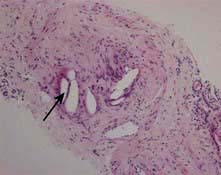
 Light chain cast nephropathy – intratubular deposition of light chains, particularly kappa chains facilitated by Tamm–Horsfall glycoprotein, which characteristically appear on renal histology as fractured casts with giant cell reaction (Fig. 12.26)
Light chain cast nephropathy – intratubular deposition of light chains, particularly kappa chains facilitated by Tamm–Horsfall glycoprotein, which characteristically appear on renal histology as fractured casts with giant cell reaction (Fig. 12.26)
AL amyloidosis – deposition of amyloid fibrils of light chains (Congo red positive)
Light chain deposition disease – nodular glomerulosclerosis with granular deposits of usually lambda light chains (Congo red negative)
Plasma cell infiltration – often incidental finding at autopsy
Fanconi’s syndrome – tubular toxicity due to light chains
Hypercalcaemic nephropathy – bone resorption causing hypercalcaemia
Hyperuricaemic nephropathy – tumour lysis causing tubular crystallization of uric acid
Radiocontrast nephropathy – interaction between light chains and radiocontrast.
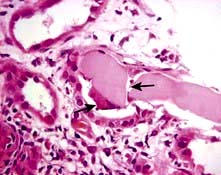
Figure 12.26 Cast nephropathy in a patient with multiple myeloma. Light microscopy picture showing characteristic fractured cast and giant cell reaction (arrows).
Treatment of underlying myeloma is indicated (p. 471). If a patient with cast nephropathy and severe AKI remains dialysis-dependent, the prognosis is poor. Commencement of effective bortezomib based chemotherapy, which decreases light chain production, and a high cut-off haemodialysis has shown some promise in relapsed myeloma.
 Box 12.3
Box 12.3