43 Anxiolytic and hypnotic drugs
Overview
In this chapter, we discuss the nature of anxiety and the drugs used to treat it (anxiolytic drugs), as well as drugs used to treat insomnia (hypnotic drugs). Historically there was overlap between these two groups, reflecting the fact that older anxiolytic drugs commonly caused a degree of sedation and drowsiness. Newer anxiolytic drugs show much less sedative effect and other hypnotic drugs have been introduced that lack specific anxiolytic effects. Many of the drugs now used to treat anxiety were first developed, and are still used, to treat other disorders such as depression and epilepsy. Here we will focus on their use as anxiolytics. Of the classical anxiolytic/hypnotic drugs, the benzodiazepines are the most important group. Possible new approaches are discussed briefly.
The Nature of Anxiety and Its Treatment
The normal fear response to threatening stimuli comprises several components, including defensive behaviours, autonomic reflexes, arousal and alertness, corticosteroid secretion and negative emotions. In anxiety states, these reactions occur in an anticipatory manner, independently of external events. The distinction between a ‘pathological’ and a ‘normal’ state of anxiety is not clear-cut but represents the point at which the symptoms interfere with normal productive activities. The term ‘anxiety’ is applied to several distinct disorders. A useful division of anxiety disorders that may help to explain why different types of anxiety respond differently to different drugs is into (i) disorders that involve fear (panic attacks and phobias) and (ii) those that involve a more general feeling of anxiety (often categorised as general anxiety disorder).
Anxiety disorders as recognised clinically include:
It should be stressed that the treatment of such disorders generally involves psychological approaches as well as drug treatment. Over the last decade the drug treatment of anxiety has changed from using traditional anxiolytic/hypnotic agents (i.e. benzodiazepines and barbiturates) to using a range of drugs that are also used to treat other CNS disorders (e.g. antidepressant, antiepileptic and antipsychotic drugs) or 5-hydroxytryptamine (5-HT)1A receptor agonists (e.g. buspirone) that have no hypnotic effect. Furthermore, benzodiazepines, while being effective anxiolytic drugs, have the disadvantages of producing unwanted side effects such as amnesia, and of inducing tolerance and physical dependence as well as being drugs of abuse. They are also ineffective in treating any depression that may occur along with anxiety. Antidepressants and buspirone do, however, require three or more weeks to show any therapeutic effect and must be taken continuously, whereas benzodiazepines can be useful for patients who need acute treatment as they reduce anxiety within 30 min, and can be taken on an ‘as needed’ basis.
Measurement of Anxiolytic Activity
Animal Models of Anxiety
In addition to the subjective (emotional) component of human anxiety, there are measurable behavioural and physiological effects that also occur in experimental animals. In biological terms, anxiety induces a particular form of behavioural inhibition that occurs in response to novel environmental events that are threatening or painful. In animals, this behavioural inhibition may take the form of immobility or suppression of a behavioural response such as bar pressing to obtain food (see below). A rat placed in an unfamiliar environment normally responds by remaining immobile although alert (behavioural suppression) for a time, which may represent ‘anxiety’ produced by the strange environment. This immobility is reduced if anxiolytic drugs are administered. The ‘elevated cross maze’ is a widely used test model. Two arms of the raised horizontal cross are closed in, and the others are open. Normally, rats spend most of their time in the closed arms and avoid the open arms (afraid, possibly, of falling off or being attacked). Administration of anxiolytic drugs increases the time spent in the open arms and also increases the number of entries made into the open arm but without an increase in motor activity.
Conflict tests can also be used. For example, a rat trained to press a bar repeatedly to obtain a food pellet normally achieves a high and consistent response rate. A conflict element is then introduced: at intervals, indicated by an auditory signal, bar pressing results in an occasional ‘punishment’ in the form of an electric shock in addition to the reward of a food pellet. Normally, the rat ceases pressing the bar (behavioural inhibition), and thus avoids the shock, while the signal is sounding. The effect of an anxiolytic drug is to relieve this suppressive effect, so that the rats continue bar pressing for reward despite the ‘punishment’. Other types of psychotropic drug are not effective, nor are analgesic drugs. Other evidence confirms that anxiolytic drugs affect the level of behavioural inhibition produced by the ‘conflict situation’, rather than simply raising the pain threshold.
Some of these ‘anxiety’ models may measure fear rather than general anxiety which occurs in humans in the absence of specific stimuli. To develop new anxiolytic drugs, it is important to have animal tests that give a good guide to efficacy in humans, and much ingenuity has gone into developing and validating such tests (see Ramos, 2008).
Tests on Humans
Various subjective ‘anxiety scale’ tests have been devised based on standard patient questionnaires. Galvanic skin reactions—a measure of sweat secretion—are also used to monitor anxiety. Neuropsychological tests have been developed to investigate emotional and attentional biases associated with responses to emotive faces and words. An experience akin to a panic attack can be induced in many subjects by breathing an increased level of CO2 (usually prolonged breathing of 7.5% CO2 or a single inhalation of 35% CO2). Such tests have confirmed the efficacy of many anxiolytic drugs, but placebo treatment often also produces highly significant responses.
A human version of the conflict test described above involves the substitution of money for food pellets, and the use of graded electric shocks as punishment. As with rats, administration of diazepam increases the rate of button pressing for money during the periods when the punishment was in operation, although the subjects reported no change in the painfulness of the electric shock.
Measurement of anxiolytic activity 
Drugs Used to Treat Anxiety
The main groups of drugs (see review by Hoffman & Mathew, 2008) are as follows:
Drugs Used to Treat Insomnia (Hypnotic Drugs)
Antidepressants (Ch. 46), antiepileptics (Ch. 44), antipsychotics Ch. 45), β-adrenoceptor antagonists (Ch. 14) and antihistamines (Ch. 26) are described in detail elsewhere in this book. Some discussion of how SSRIs exert their anxiolytic activity is included in the section on buspirone (see below). In this chapter we focus on drugs whose primary use is as anxiolytic and hypnotic agents.
Classes of anxiolytic and hypnotic drugs
Benzodiazepines and Related Drugs
 The first benzodiazepine, chlordiazepoxide, was synthesised by accident in 1961, the unusual seven-membered ring having been produced as a result of a reaction that went wrong in the laboratories of Hoffman-La Roche. Its unexpected pharmacological activity was recognised in a routine screening procedure, and benzodiazepines quite soon became the most widely prescribed drugs in the pharmacopoeia.
The first benzodiazepine, chlordiazepoxide, was synthesised by accident in 1961, the unusual seven-membered ring having been produced as a result of a reaction that went wrong in the laboratories of Hoffman-La Roche. Its unexpected pharmacological activity was recognised in a routine screening procedure, and benzodiazepines quite soon became the most widely prescribed drugs in the pharmacopoeia.
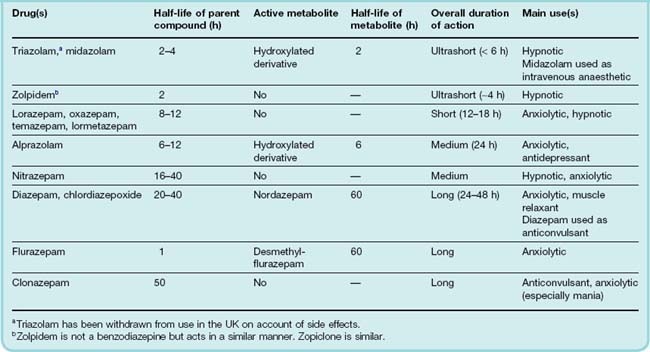
The basic chemical structure of benzodiazepines consists of a seven-membered ring fused to an aromatic ring, with four main substituent groups that can be modified without loss of activity. Thousands of compounds have been made and tested, and about 20 are available for clinical use, the most important ones being listed in Table 43.1. They are basically similar in their pharmacological actions, although some degree of selectivity has been reported. For example, some, such as clonazepam, show anticonvulsant activity with less marked sedative effects. From a clinical point of view, differences in pharmacokinetic behaviour among different benzodiazepines (see below) are more important than differences in profile of activity. Drugs with a similar structure have been discovered that specifically antagonise the effects of the benzodiazepines, for example flumazenil (see below).
The term ‘benzodiazepine’ refers to a distinct chemical structure. Drugs such as zolpidem and zopiclone have a different chemical structure and are therefore not benzodiazepines. However, since they bind to the same sites, often referred to as the ‘benzodiazepine receptor’, they are discussed along with the benzodiazepines.
Mechanism of Action
Benzodiazepines act selectively on GABAA receptors (Ch. 37), which mediate inhibitory synaptic transmission throughout the central nervous system. Benzodiazepines enhance the response to GABA by facilitating the opening of GABA-activated chloride channels (Fig. 37.4). They bind specifically to a regulatory site on the receptor, distinct from the GABA-binding sites (see below), and act allosterically to increase the affinity of GABA for the receptor. Single-channel recordings show an increase in the frequency of channel opening by a given concentration of GABA, but no change in the conductance or mean open time, consistent with an effect on GABA binding rather than the channel-gating mechanism. Benzodiazepines do not affect receptors for other amino acids, such as glycine or glutamate (Fig. 43.1).
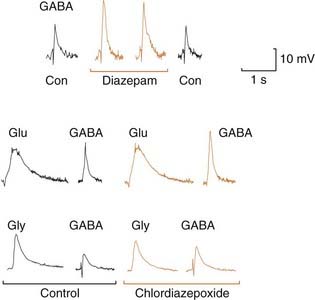
Fig. 43.1 Potentiating effect of benzodiazepines and chlordiazepoxide on the action of GABA.
Drugs were applied by ionophoresis to mouse spinal cord neurons grown in tissue culture, from micropipettes placed close to the cells. The membrane was hyperpolarised to −90 mV, and the cells were loaded with Cl− from the recording microelectrode, so inhibitory amino acids (GABA and glycine, Gly), as well as excitatory ones (glutamate, Glu), caused depolarising responses. The potentiating effect of diazepam is restricted to GABA responses, glutamate and glycine responses being unaffected. Con, control.
The GABAA receptor is a ligand-gated ion channel (see Ch. 3) consisting of a pentameric assembly of different subunits, the main ones being α, β and γ (see Ch. 37). The GABAA receptor should actually be thought of as a family of receptors as there are six different subtypes of α subunit, three subtypes of β and three subtypes of γ. Although the potential number of combinations is therefore large, certain combinations predominate in the adult brain (see Ch. 37). The various combinations occur in different parts of the brain, have different physiological functions and have subtle differences in their pharmacological properties (see below).
Benzodiazepines bind across the interface between the α and γ subunits but only to receptors that contain γ2 and α1, α2, α3 or α5 subunits. Two genetic approaches have been used to study the roles of different subunits in the different behavioural effects of benzodiazepines—genetic knockout and loss of function mutants (see Whiting, 2003; Reynolds, 2008). The loss of function mutant approach has the advantage over subunit knockout that it reduces the likelihood of compensatory changes in the expression of other subunits. Mutation of a single amino acid (histidine 101 or its equivalent) in the α subunit eliminates benzodiazepine binding. Behavioural analysis of various mutant mice indicates that α1-containing receptors mediate the sedative but not the anxiolytic effect of benzodiazepines whereas α2- and α3-containing receptors mediate the anxiolytic effect.
The obvious next step has been to try and develop subunit-selective drugs (Reynolds, 2008; Christmas et al., 2008). Unfortunately, this has proved difficult, due to the structural similarity between the benzodiazepine binding site on different α subunits. What has been possible is the development of drugs that, while having little subunit-selectivity of binding, have different levels of agonist efficacy at receptors containing different subunits. Selective efficacy at α2- and α3-containing receptors may produce drugs that have an anxiolytic effect without the unwanted effects of sedation and amnesia. Such compounds have been developed (e.g. MK-0343, TPA023) but only limited data on their effectiveness in humans are currently available. Pagoclone, which is reported to be an α3 agonist and α1, α2 and α5 partial agonist, has little or no sedative or amnesic actions and is in development for the treatment of panic disorders and stuttering.
Peripheral benzodiazepine-binding sites, not associated with GABA receptors, are known to exist in many tissues. They are located primarily on mitochondrial membranes. For information on their structure and functions, see Veenman & Gavish (2006).
Pharmacological Effects and Uses
The main effects of benzodiazepines are:
Reduction of anxiety and aggression
Benzodiazepines show anxiolytic effects in animal tests, as described above, and also exert a marked ‘taming’ effect, allowing animals to be handled more easily.3 If given to the dominant member of a pair of animals (e.g. mice or monkeys) housed in the same cage, benzodiazepines reduce the number of attacks by the dominant individual and increase the number of attacks made on him. With the possible exception of alprazolam (Table 43.1), benzodiazepines do not have antidepressant effects. Benzodiazepines may paradoxically produce an increase in irritability and aggression in some individuals. This appears to be particularly pronounced with the ultrashort-acting drug triazolam (and led to its withdrawal in the UK and some other countries), and is generally more common with short-acting compounds. It is probably a manifestation of the benzodiazepine withdrawal syndrome, which occurs with all these drugs (see below) but is more acute with drugs whose action wears off rapidly.
Benzodiazepines are now used mainly for treating acute anxiety states.
Induction of sleep and sedation
Benzodiazepines decrease the time taken to get to sleep, and increase the total duration of sleep, although the latter effect occurs only in subjects who normally sleep for less than about 6 h each night. With agents that have a short duration of action (e.g. zolpidem or temazepam), a pronounced hangover effect on wakening can be avoided.
On the basis of electroencephalography measurements, several levels of sleep can be recognised. Of particular psychological importance are rapid-eye-movement (REM) sleep, which is associated with dreaming, and slow-wave sleep, which corresponds to the deepest level of sleep when the metabolic rate and adrenal steroid secretion are at their lowest and the secretion of growth hormone is at its highest (see Ch. 32). Most hypnotic drugs reduce the proportion of REM sleep, although benzodiazepines affect it less than other hypnotics, and zolpidem (see below) least of all. Artificial interruption of REM sleep causes irritability and anxiety, even if the total amount of sleep is not reduced, and the lost REM sleep is made up for at the end of such an experiment by a rebound increase. The same rebound in REM sleep is seen at the end of a period of administration of benzodiazepines or other hypnotics. The proportion of slow-wave sleep is significantly reduced by benzodiazepines, although growth hormone secretion is unaffected.
Figure 43.2 shows the improvement of subjective ratings of sleep quality produced by a benzodiazepine, and the rebound decrease at the end of a 32-week period of drug treatment. It is notable that, although tolerance to objective effects such as reduced sleep latency occurs within a few days, this is not obvious in the subjective ratings.
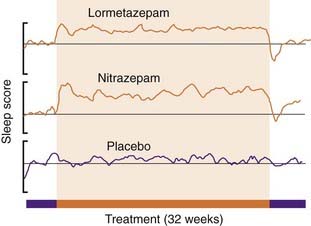
Fig. 43.2 Effects of long-term benzodiazepine treatment on sleep quality.
A group of 100 poor sleepers were given, under double-blind conditions, lormetazepam 5 mg, nitrazepam 2 mg or placebo nightly for 24 weeks, the test period being preceded and followed by 4 weeks of placebo treatment. They were asked to assess, on a subjective rating scale, the quality of sleep during each night, and the results are expressed as a 5-day rolling average of these scores. The improvement in sleep quality was maintained during the 24-week test period, and was followed by a ‘rebound’ worsening of sleep when the test period ended.
(From Oswald I et al. 1982 Br Med J 284: 860–864.)
Benzodiazepines are now, however, only recommended for short courses of treatment of insomnia. Tolerance develops over 1–2 weeks with continuous use, and on cessation rebound insomnia and a withdrawal syndrome may occur (see below).
Benzodiazepines are also used as premedication before surgery (both medical and dental). Under these circumstances their anxiolytic, sedative and amnesic properties may be beneficial. Intravenous midazolam can be used to induce anaesthesia (see Ch. 40).
Reduction of muscle tone
Benzodiazepines reduce muscle tone by a central action on GABAA receptors primarily in the spinal cord.
Increased muscle tone is a common feature of anxiety states in humans and may contribute to the aches and pains, including headache, which often trouble anxious patients. The relaxant effect of benzodiazepines may therefore be clinically useful. A reduction of muscle tone appears to be possible without appreciable loss of coordination. However, with intravenous administration in anaesthesia and in overdose when these drugs are being abused, airway obstruction may occur. Other clinical uses of muscle relaxants are discussed in Chapter 13.
Anticonvulsant effects
All the benzodiazepines have anticonvulsant activity in experimental animal tests. They are highly effective against chemically induced convulsions caused by pentylenetetrazol, bicuculline and similar drugs that act by blocking GABAA receptors (see Chs 37 and 44) but less so against electrically induced convulsions.
Clonazepam (see above) is used to treat epilepsy (Ch. 44), as is diazepam, which is administered rectally to children in acute seizures and intravenously to control life-threatening seizures in status epilepticus. Tolerance develops to the anticonvulsant actions of benzodiazepines (see below).
Anterograde amnesia
Benzodiazepines prevent memory of events experienced while under their influence, an effect not seen with other CNS depressants. Minor surgical or invasive procedures can thus be performed without leaving unpleasant memories. Flunitrazepam (better known to the general public by one of its trade names, Rohypnol) is infamous as a date rape drug and victims frequently have difficulty in recalling exactly what took place during the attack.
Amnesia is thought to be due to benzodiazepines binding to GABAA receptors containing the α5 subunit. α5 Knockout mice show an enhanced learning and memory phenotype. This raises the possibility that an α5-subunit-selective inverse agonist (see below for a general description of benzodiazepine inverse agonism) could be memory enhancing.
Is There an Endogenous Benzodiazepine-Like Mediator?
Despite considerable scientific effort, the question of whether or not there are endogenous ligands for the benzodiazepine receptors, whose function is to regulate the action of GABA, remains unanswered.
That the antagonist flumazenil produces responses both in vivo and in vitro in the absence of any exogenous benzodiazepines is frequently cited to support the view that there must be ongoing benzodiazepine receptor activation by endogenous ligand(s). Although flumazenil was originally described as a neutral antagonist (see below), it is possible, however, that it has agonist or inverse agonist activity at subtypes of GABAA receptor (depending on the α subunit present) or in some pathological conditions in which the GABAA receptors have become modified.
Several molecules that act on benzodiazepine receptors have been isolated, including β-carbolines (e.g. ethyl-β-carboline-3-carboxylate, βCCE), structurally related to tryptophan, and diazepam-binding inhibitor, a 10-kDa peptide. Whether these molecules exist in the brain or are generated during the processes involved in extracting them from the tissue is an open issue. Interestingly both βCCE and diazepam-binding inhibitor have the opposite effect to benzodiazepines, i.e. they are inverse agonists and inhibit chloride channel opening by GABA and, in the whole animal, exert anxiogenic and proconvulsant effects. There was also a suggestion that benzodiazepines themselves may occur naturally in the brain but the origin of these compounds and how biosynthesis occurs is unclear. At present, there is no general agreement on the identity and function of endogenous ligands for the benzodiazepine receptor. Other possible endogenous modulators of GABAA receptors include steroid metabolites but they bind to a different site from benzodiazepines (see Ch. 40).
Pharmacokinetic Aspects
Benzodiazepines are well absorbed when given orally, usually giving a peak plasma concentration in about 1 h. Some (e.g. oxazepam, lorazepam) are absorbed more slowly. They bind strongly to plasma protein, and their high lipid solubility causes many of them to accumulate gradually in body fat. They are normally given by mouth but can be given intravenously (e.g. diazepam in status epilepticus, midazolam in anaesthesia). Intramuscular injection often results in slow absorption.
Benzodiazepines are all metabolised and eventually excreted as glucuronide conjugates in the urine. They vary greatly in duration of action and can be roughly divided into short-, medium- and long-acting compounds (Table 43.1). Duration of action influences their use, short-acting compounds being useful hypnotics with reduced hangover effect on wakening, long-acting compounds being more useful for use as anxiolytic and anticonvulsant drugs. Several are converted to active metabolites such as N-desmethyldiazepam (nordazepam), which has a half-life of about 60 h, and which accounts for the tendency of many benzodiazepines to produce cumulative effects and long hangovers when they are given repeatedly. The short-acting compounds are those that are metabolised directly by conjugation with glucuronide. Figure 43.3 shows the gradual build up and slow disappearance of nordazepam from the plasma of a human subject given diazepam daily for 15 days.
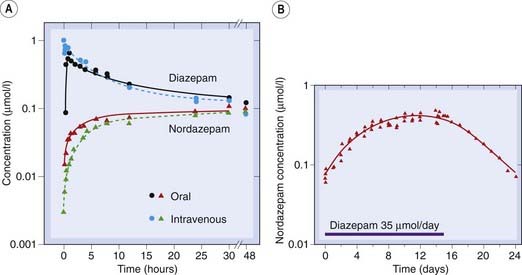
Fig. 43.3 Pharmacokinetics of diazepam in humans.
[A] Concentrations of diazepam and nordazepam following a single oral or intravenous dose. Note the very slow disappearance of both substances after the first 20 h. [B] Accumulation of nordazepam during 2 weeks’ daily administration of diazepam, and slow decline (half-life about 3 days) after cessation of diazepam administration.
(Data from Kaplan S A et al. 1973 J Pharmacol Sci 62: 1789.)
Advancing age affects the rate of oxidative reactions more than that of conjugation reactions. Thus the effect of the long-acting benzodiazepines tends to increase with age, and it is common for drowsiness and confusion to develop insidiously for this reason.4
Unwanted Effects
These may be divided into:
Acute toxicity
Benzodiazepines in acute overdose are considerably less dangerous than other anxiolytic/hypnotic drugs. Because such agents are often used in attempted suicide, this is an important advantage. In overdose, benzodiazepines cause prolonged sleep, without serious depression of respiration or cardiovascular function. However, in the presence of other CNS depressants, particularly alcohol, benzodiazepines can cause severe, even life-threatening, respiratory depression. This is a frequent problem when benzodiazepines are used as recreational drugs (see Chs 48 and 58). The availability of an effective antagonist, flumazenil, means that the effects of an acute overdose can be counteracted,5 which is not possible for most CNS depressants.
Side effects during therapeutic use
The main side effects of benzodiazepines are drowsiness, confusion, amnesia and impaired coordination, which considerably affects manual skills such as driving performance. Benzodiazepines enhance the depressant effect of other drugs, including alcohol, in a more than additive way. The long and unpredictable duration of action of many benzodiazepines is important in relation to side effects. Long-acting drugs such as nitrazepam are no longer used as hypnotics, and even shorter-acting compounds such as lorazepam can produce a substantial day-after impairment of job performance and driving skill.
Tolerance and dependence
Tolerance (i.e. a gradual escalation of dose needed to produce the required effect) occurs with all benzodiazepines, as does dependence, which is their main drawback. They share these properties with other sedatives. Tolerance appears to represent a change at the receptor level, but the mechanism is not well understood (Wafford, 2005).
At the receptor level, the degree of tolerance will be governed both by the number of receptors occupied (i.e. the dose) and the duration of receptor occupancy (which may vary according to the therapeutic use). Therefore, marked tolerance develops when benzodiazepines are used continuously to treat epilepsy whereas less tolerance occurs to the sleep-inducing effect when the subject is relatively drug free during the day. It is not clear to what degree tolerance develops to the anxiolytic effect.
Benzodiazepines produce dependence, and this is a major problem. In human subjects and patients, abrupt cessation of benzodiazepine treatment after weeks or months causes a rebound heightened anxiety, together with tremor, dizziness, tinnitus, weight loss and disturbed sleep due to enhanced REM sleep. It is recommended that benzodiazepines be withdrawn gradually by stepwise lowering of the dose. Although animals show only a weak tendency to self-administer benzodiazepines, withdrawal after chronic administration causes physical symptoms, namely nervousness, tremor, loss of appetite and sometimes convulsions.6 The withdrawal syndrome, in both animals and humans, is slower in onset than with opioids, probably because of the long plasma half-life of most benzodiazepines. With diazepam, the withdrawal symptoms may take up to 3 weeks to become apparent. Short-acting benzodiazepines cause more abrupt withdrawal effects. With triazolam, a very short-acting drug and no longer in use, the withdrawal effect occurred within a few hours, even after a single dose, producing early-morning insomnia and daytime anxiety when the drug was used as a hypnotic.
The physical and psychological withdrawal symptoms make it difficult for patients to give up taking benzodiazepines, but craving (i.e. severe psychological dependence that outlasts the physical withdrawal syndrome), which occurs with many drugs of abuse (Ch. 48), is not a major problem.
Benzodiazepine Antagonists and Inverse Agonists
Competitive antagonists of benzodiazepines were first discovered in 1981. The best-known compound is flumazenil. This compound was originally reported to lack effects on behaviour or on drug-induced convulsions when given on its own, although it was later found to possess some ‘anxiogenic’ and proconvulsant activity. Flumazenil can be used to reverse the effect of benzodiazepine overdosage (normally used only if respiration is severely depressed), or to reverse the effect of benzodiazepines such as midazolam used for minor surgical procedures. Flumazenil acts quickly and effectively when given by injection, but its action lasts for only about 2 h, so drowsiness tends to return. It can be used to treat comatose patients suspected of having overdosed with benzodiazepines. Convulsions may rarely occur in patients treated with flumazenil, and this is more common in patients receiving tricyclic antidepressants (Ch. 46). Reports that flumazenil improves the mental state of patients with severe liver disease (hepatic encephalopathy) and alcohol intoxication have not been confirmed in controlled trials although partial inverse agonists do appear to be effective in animal models of hepatic encephalopathy (Ahboucha & Butterworth, 2005).
The term inverse agonist (Ch. 2) is applied to drugs that bind to benzodiazepine receptors and exert the opposite effect to that of conventional benzodiazepines, producing signs of increased anxiety and convulsions. βCCE, diazepam-binding inhibitor (see above) and some benzodiazepine analogues show inverse agonist activity. It is possible (see Fig. 43.4) to explain these complexities in terms of the two-state model discussed in Chapter 2, by postulating that the benzodiazepine receptor exists in two distinct conformations, only one of which (A) can bind GABA molecules and open the chloride channel. The other conformation (B) cannot bind GABA. Normally, with no benzodiazepine receptor ligand present, there is an equilibrium between these two conformations; sensitivity to GABA is present but submaximal. Benzodiazepine agonists (e.g. diazepam) are postulated to bind preferentially to conformation A, thus shifting the equilibrium in favour of A and enhancing GABA sensitivity. Inverse agonists bind selectively to B and have the opposite effect. Competitive antagonists would bind equally to A and B, and consequently would not disturb the conformational equilibrium but antagonise the effect of both agonists and inverse agonists
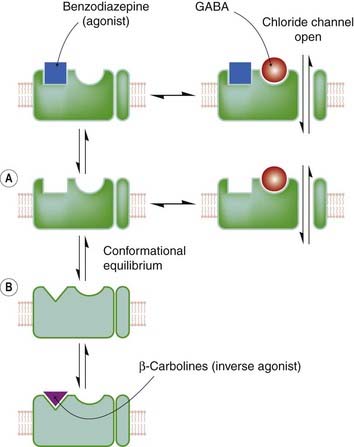
Fig. 43.4 Model of benzodiazepine/GABA receptor interaction.
Benzodiazepine agonists, antagonists and inverse agonists are believed to bind to a site on the GABA receptor distinct from the GABA-binding site. A conformational equilibrium exists between states in which the benzodiazepine receptor exists in its agonist-binding conformation (A), and in its inverse agonist-binding conformation (B). In the latter state, the GABA receptor has a much reduced affinity for GABA; consequently, the chloride channel remains closed.
Benzodiazepines
Buspirone
Buspirone is used to treat generalised anxiety disorders. It is ineffective in controlling panic attacks or severe anxiety states.
Buspirone is a partial agonist at 5-HT1A receptors (Ch. 15) and also binds to dopamine receptors, but it is likely that its 5-HT-related actions are important in relation to anxiety suppression, because related experimental compounds (e.g. ipsapirone and gepirone) which are highly specific for 5-HT1A receptors, show similar anxiolytic activity in experimental animals (see Traber & Glaser, 1987). However, buspirone takes days or weeks to produce its effect in humans, suggesting a more complex mechanism of action than simply activation of 5-HT1A receptors. SSRIs also have a delayed onset to their anxiolytic actions.
5-HT1A receptors are expressed on the soma and dendrites of 5-HT-containing neurons, where they function as inhibitory autoreceptors, as well as being expressed on other types of neuron (e.g. noradrenergic locus coeruleus neurons) where, along with other types of 5-HT receptor (see Ch. 38), they mediate the postsynaptic actions of 5-HT. Postsynaptic 5-HT1A receptors are highly expressed within the cortico-limbic circuits implicated in emotional behaviour. One theory of how buspirone and SSRIs produce their delayed anxiolytic effect is that over time they induce desensitisation of somatodendritic 5-HT1A autoreceptors resulting in heightened excitation of serotonergic neurons and enhanced 5-HT release. This might also explain why early in treatment anxiety can be worsened by these drugs due to the initial activation of 5-HT1A autoreceptors and inhibition of 5-HT release. This receptor desensitisation theory would predict that a 5-HT1A antagonist that would rapidly block the action of 5-HT at 5-HT1A autoreceptors and thus swiftly enhance 5-HT release might be anxiolytic without delayed onset. Drugs with combined 5-HT1A antagonism and SSRI properties have been developed but have not been found to be effective in man, perhaps because they block both 5HT1A autoreceptors and postsynaptic receptors, the latter effect occluding the beneficial effect of the former. Elevated 5-HT levels may also induce other postsynaptic adaptations. Receptors that have received particular interest are the 5-HT2 receptors and downregulation of these may be important for anxiolytic action. Drugs with 5-HT2 and 5-HT3 receptor antagonist activity are in clinical trials for treating anxiety.
Buspirone inhibits the activity of noradrenergic locus coeruleus neurons (Ch. 38) and thus interferes with arousal reactions. It has side effects quite different from those of benzodiazepines. It does not cause sedation or motor incoordination, nor have tolerance or withdrawal effects been reported. Its main side effects are nausea, dizziness, headache and restlessness, which generally seem to be less troublesome than the side effects of benzodiazepines. Buspirone does not suppress the benzodiazepine withdrawal syndrome, presumably because it acts by a different mechanism. Hence, when switching from benzodiazepine treatment to buspirone treatment, the benzodiazepine dose still needs to be reduced gradually (see above).
Antidepressants and 5-HT1A agonists as anxiolytic drugs
Other Potential Anxiolytic Drugs
Besides the GABA and 5-HT mechanisms discussed above, many other transmitters and hormones have been implicated in anxiety and panic disorders, particularly noradrenaline, glutamate, corticotrophin-releasing factor, cholecystokinin (CCK), substance P, neuropeptide Y, galanin, orexins and neurosteroids. Anxiolytic drugs aimed at these targets are in development, but none is so far available for clinical use (see Christmas et al., 2008; Mathew et al., 2009).
Clinical use of drugs as anxiolytics 
Clinical use of hypnotics (‘sleeping tablets’)
References and Further Reading
Ahboucha S., Butterworth R.F. Role of endogenous benzodiazepine ligands and their GABAA-associated receptors in hepatic encephalopathy. Metab. Brain. Dis.. 2005;20:425-437.
Christmas D., Hood S., Nutt D. Potential novel anxiolytic drugs. Curr. Pharm. Des.. 2008;14:3534-3546. (Explains brain mechanisms thought to underlie actions of anxiolytic drugs)
Hoffman E.J., Mathew S.J. Anxiety disorders: a comprehensive review of pharmacotherapies. Mt. Sinai. J. Med.. 2008;75:248-262. (Describes the clinical usefulness of various drugs effective against different forms of anxiety)
Mathew S.J., Price R.B., Charney D.S. Recent advances in the neurobiology of anxiety disorders: implications for novel therapeutics. Am. J. Med. Genet. C. Semin. Med. Genet.. 2009;148C:89-98. (This review focuses on the potential for development of novel treatments for anxiety)
Ramos A. Animal models of anxiety: do I need multiple tests? Trends. Pharmacol. Sci.. 2008;29:493-498. (Describes the need for animal models in the testing of anxiolytic drugs)
Reynolds D.S. The value of genetic and pharmacological approaches to understanding the complexities of GABAA receptor subtype functions: the anxiolytic effects of benzodiazepines Pharmacol. Biochem. Behav. 90:2008:37-42 (Describes recent work with transgenic mice expressing mutated GABAA receptors, suggesting that anxiolytic and sedative actions of benzodiazepines may be separable)
Traber J., Glaser T. 5-HT1A receptor-related anxiolytics. Trends. Pharmacol. Sci.. 1987;8:432-437.
Veenman L., Gavish M. The peripheral-type benzodiazepine receptor and the cardiovascular system. Implications for drug development. Pharmacol. Ther.. 2006;110:503-524. (Useful update on peripheral benzodiazepine receptors in mitochondria)
Wafford K.A. GABAA receptor subtypes:any clues to the mechanism of benzodiazepine dependence? Curr. Opin. Pharmacol.. 2005;5:47-52.
Whiting P. GABAA receptor subtypes in the brain: a paradigm for CNS drug discovery? Drug. Discov. Today 8:2003:445-450 (Useful summary of the extensive data relating to GABAA receptor subtypes in relation to benzodiazepine and anaesthetic pharmacology)
1β-Blockers are sometimes used by actors and musicians to reduce the symptoms of stage fright, but their use by snooker players to minimise tremor is banned as unsportsmanlike.
2This is an interesting example of an initial unwanted side effect—sedation is undesired when treating hay fever—subsequently becoming a therapeutic use.
3This depends on the species. Cats actually become more excitable, as a colleague of one of the authors discovered to his cost when attempting to sedate a tiger in the Baltimore zoo.
4At the age of 91, the grandmother of one of the authors was growing increasingly forgetful and mildly dotty, having been taking nitrazepam for insomnia regularly for years. To the author’s lasting shame, it took a canny general practitioner to diagnose the problem. Cancellation of the nitrazepam prescription produced a dramatic improvement.
5In practice, patients are usually left to sleep it off, because there is a risk of seizures with flumazenil; however, flumazenil may be useful diagnostically to rule out coma of other causes.
6Withdrawal symptoms can be more severe. A relative of one of the authors, advised to stop taking benzodiazepines after 20 years, suffered hallucinations and one day tore down all the curtains, convinced that they were on fire.