Disorders of the Gastrointestinal Tract
Congenital defects of the GI tract can involve any portion from the mouth to the anus. Most are apparent at birth or shortly thereafter and are anomalies in which normal growth ceased at a crucial stage of embryonic development, leaving the structure in an embryonic form or only partially completed. The result may be atresia, malposition, nonclosure, or any number of variations.
Atresia is absence of a normal opening or normally patent lumen. Atresia at any point along the length of the GI tract creates an obstruction to the normal progress of nutrients and secretions. The most common anomalies requiring surgical intervention are atresias of the esophagus, intestine, and anus. The congenital defects considered in this chapter include abnormalities of the lip and palate, esophagus, and anus. Some malformations of the GI tract are considered here rather than in Chapter 33 because they are identified at birth and are cause for considerable parental concern.
Cleft Lip and Cleft Palate
 Cleft lip (CL) with or without cleft palate (CP) is the most common birth defect in the United States and occurs with a frequency of 1 in 600 live births (Cleft Palate Foundation, 2008). Isolated CP has an incidence of approximately 1 in 2500 live births (Tinanoff, 2007). CL with or without CP is more common in males, and CP alone is more common in females. CL appears more often in Native Americans and Asians and less frequently in African-Americans. Although the majority of clefts are nonsyndromic (have no associated identifiable syndrome), associated syndromes occur in varying frequencies according to the specific defect. It is estimated that 10% to 50% of children with CL/CP have an associated syndrome (Arosarena, 2007; Merritt, 2005). See Table 11-5 for a comparison of the two defects.
Cleft lip (CL) with or without cleft palate (CP) is the most common birth defect in the United States and occurs with a frequency of 1 in 600 live births (Cleft Palate Foundation, 2008). Isolated CP has an incidence of approximately 1 in 2500 live births (Tinanoff, 2007). CL with or without CP is more common in males, and CP alone is more common in females. CL appears more often in Native Americans and Asians and less frequently in African-Americans. Although the majority of clefts are nonsyndromic (have no associated identifiable syndrome), associated syndromes occur in varying frequencies according to the specific defect. It is estimated that 10% to 50% of children with CL/CP have an associated syndrome (Arosarena, 2007; Merritt, 2005). See Table 11-5 for a comparison of the two defects.
 Case Study—Cleft Lip and Palate
Case Study—Cleft Lip and Palate
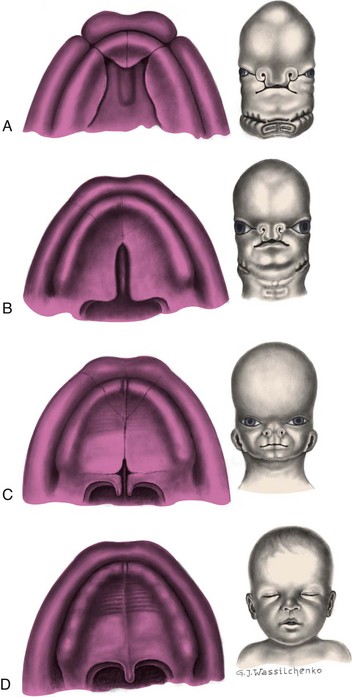
CL results from incomplete fusion of the embryonic structures surrounding the primitive oral cavity. The cleft may be unilateral or bilateral and is often associated with abnormal development of the external nose, nasal cartilages, nasal septum, and maxillary alveolar ridges. It may or may not be associated with CP. The extent of the cleft varies greatly from an indentation in the lip to a deep and wide fissure extending to the nostril. In CL, there is nasal slumping with collapse of the alar dome on the affected side. The columella is deviated to the unaffected side, pulling the nasal tip in that direction. In bilateral CL, the prolabium may be partially or completely separated from the lateral portion of the upper lip. This may extend into the gumline, separating the premaxilla from the remainder of the alveolar ridge. The premaxilla may deviate anteriorly outside of the oral cavity. Dental anomalies, such as missing, malpositioned, or deformed primary teeth, are common at the site of the cleft. Secondary teeth may or may not be affected, but will need bone in the alveolar cleft area in which to anchor during eruption.
CP occurs when the primary and secondary palatine plates fail to fuse during embryonic development. CPs vary greatly in degree and may involve only the soft palate or may extend into the hard palate. The cleft may occur on one side (unilateral) or both sides (bilateral). It can be incomplete or complete, or a combination of the two, independent of clefting of the lip. Wide central palatal clefts, often described as “horseshoe-shaped” clefts, may be associated with PRS. These children have associated micrognathia (retracted mandible) and airway issues related to the posterior positioning of the tongue. The cleft may occur only in the midline of the posterior palate or may extend to the nostril on one or both sides. When the lip is unattached and displaced forward, a portion of the alveolus is similarly detached. Occasionally, small clefts of the soft palate may be difficult to identify. A submucous CP may also be difficult to identify initially because the palatal cleft is covered by the mucous membrane of the roof of the mouth. Classic stigmata of the submucous cleft include a bifid uvula, a bony notch in the hard palate, and a zona pellucida (a blue or whitish line that courses the midline of the soft palate). The submucous CP may be associated with speech problems and hypernasality in some cases. A bifid uvula, the smallest form of a velar cleft, is not in itself associated with speech or feeding problems.
Etiology
Many factors appear to be involved in the etiology of CL and CP, and evidence indicates that CL with or without CP is developmentally and genetically different from isolated CP. The majority of cases appear to be consistent with the concept of multifactorial inheritance as evidenced by an increased incidence in relatives and a higher concordance in monozygotic twins than in dizygotic twins. Siblings of children with CL with or without CP have an increased risk of the same anomaly but not of CP alone.
Many recognized syndromes include CL and CP as a feature. Some of these syndromes are a result of chromosomal abnormalities, and environmental factors or teratogens may be responsible for clefts at a critical point in embryonic development. Drugs such as phenytoin, valproic acid, thalidomide, and the pesticide dioxin can cause CL/CP. Maternal nutrition, especially folic acid deficiency, has been linked to clefting in humans, as have maternal alcohol ingestion and smoking during pregnancy. Alcohol consumption, specifically binge drinking (more than five drinks per sitting), increases the risk of having an infant with a cleft (DeRoo, Wilcox, Drevon, et al, 2008). Evidence shows that maternal smoking early in pregnancy is associated with a 1.5- to 2-fold increase in the risk for orofacial clefts, especially isolated clefts, with the risk increasing proportionately with the number of cigarettes smoked (Little, Cardy, and Munger, 2004).
Pathophysiology
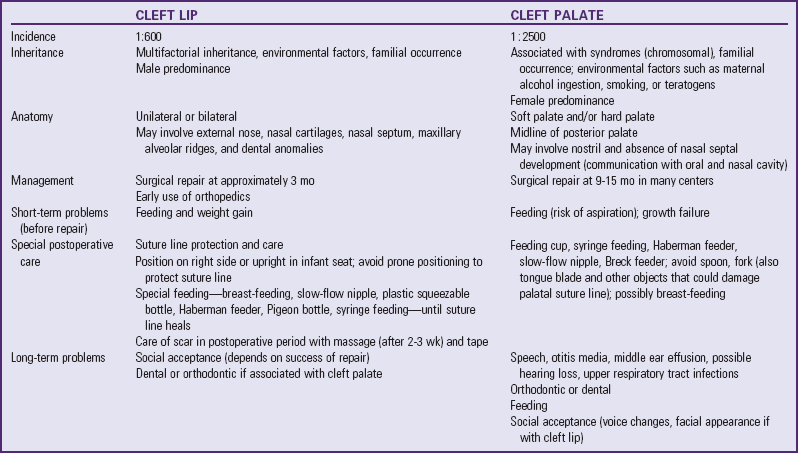
Development of the primary and secondary palates takes place at different times and involves different developmental processes. CL, or the primary palate, includes the upper lip and extends through the alveolar ridge. CP, or the secondary palate, starts posterior to the alveolar ridge and extends through the uvula. CL with or without CP results from failure of the maxillary processes to fuse with the nasal elevations on the frontal prominence, which normally occurs during the sixth week of gestation (Fig. 11-15, A). In some cases CP may occur as a result of a rupture of unstable mesoderm layer resulting in a cleft. Merging of the upper lip at the midline is completed between the seventh and eighth weeks of gestation.
Fusion of the secondary palate (hard and soft palates) takes place later in development, between the seventh and twelfth weeks of gestation (Fig. 11-15, B to D). At the time the primary palate is completed, the two lateral palatine processes are situated in a vertical position at the side of the tongue. In the process of migrating to a horizontal position, they are, for a short time, separated by the tongue. With development of the neck and jaws, the tongue moves downward, allowing the palatine processes to fuse with one another and with the primary palate to form the roof of the mouth. If there is delay in this movement, or if the tongue fails to descend soon enough, the remainder of development proceeds but the palate never fuses.
Diagnostic Evaluation
A cleft that involves the lip with or without CP is readily apparent at birth and is a defect that may elicit significant distress in parents.
Palpation of the hard palate and soft palate, submucous palate, and uvula with a gloved finger and visual inspection of the oral cavity and its structures are important parts of the newborn physical examination. (See Chapter 8.) The degree of malformation of the CL or CP can then be evaluated (Figs. 11-16 and 11-17). Clefts of the lip may be unilateral or bilateral, and the extent of the cleft and degree of nasal deformity vary. As with CL, the degree of deformity with CP varies and may involve only the uvula or may extend through both the soft and hard palates. The severity of the CP has an impact on feeding problems; the infant is unable to generate negative pressure and create suction in the oral cavity. This impairs feeding even though in most cases the infant’s ability to swallow is normal.
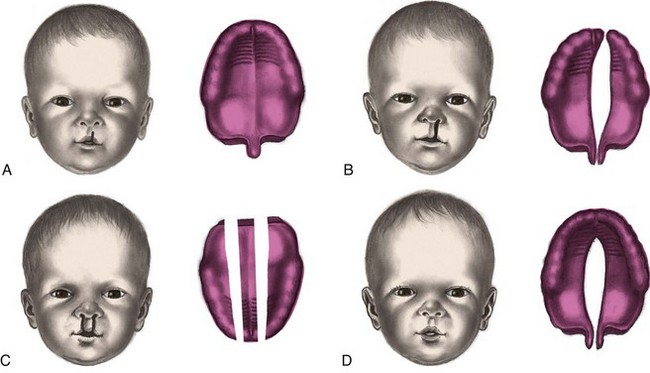
Fig. 11-16 Variations in clefts of lip and palate at birth. A, Notch in vermilion border. B, Unilateral cleft lip and cleft palate. C, Bilateral cleft lip and cleft palate. D, Cleft palate.
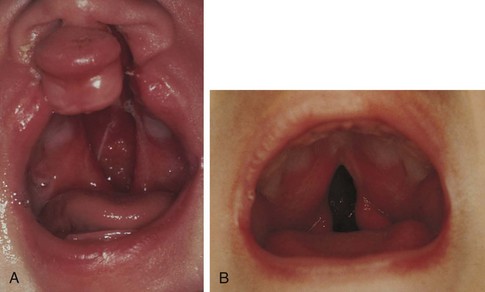
Fig. 11-17 A, Bilateral cleft lip with complete cleft palate. Cleft extends from soft to hard palate, exposing nasal cavity. B, Midline cleft of soft palate. (From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 5, St Louis, 2007, Mosby; B, Courtesy Barbara Elster, Cleft Palate Center, Pittsburgh.)
Therapeutic Management
Treatment of the child with isolated CL is surgical and involves the craniofacial multidisciplinary team. Depending on the severity of the CL, the team may include the surgeon, dentist or orthodontist, speech-language pathologist, nurse, and social worker. Management of CP also involves the cooperative efforts of a multidisciplinary health care team, including pediatrics, plastic surgery, orthodontics, otolaryngology, speech and language pathology, audiology, nursing, and social work. Treatment continues over a long time, but, even after completion of a program of health care, the child may retain defects of speech, facial appearance, and other problems related to the cleft. Management of both defects is directed toward surgical closure of the cleft, prevention of complications, and facilitation of normal growth and development of the child.
Surgical Correction: Cleft Lip: Closure of the lip defect precedes that of the palate, usually during the first few months of life. Many surgeons prefer the child to be 10 weeks of age and weigh 4.5 kg (10 lb); however, this varies. An important consideration in the timing of the surgery is the size of the cleft. Surgical correction is performed when the infant is free of any oral, respiratory, or systemic infection. Repair of the CL involves a rotation advancement flap, resulting in a scar that mimics the philtrum column.
Improved surgical techniques have minimized deformity related to scar retraction, but optimum cosmetic results are difficult to obtain in severe defects. CL healing takes place with minimal scar formation. Optimal care in the postoperative period includes scar massage (approximately 2 weeks postoperatively) and the use of silicone tape and gels several weeks postoperatively. Remaining physical characteristics in the older child are residual nasal deformity, a mildly protruding lower lip secondary to midfacial hypoplasia, and a somewhat flattened lower third of the upper lip, usually with an abnormally shaped vermillion. Not infrequently, revisions may be required at a later age.
Surgical Correction: Cleft Palate: CP repair was previously postponed until a later age than repair of the CL to take advantage of palatal changes that occur with normal growth. However, the timing of repairs remains controversial and is typically performed at 9 to 15 months in most centers to maximize speech production and growth of the midface (Arosarena, 2007). Most CP teams prefer to close the cleft before the child develops compensatory speech patterns. Recent studies have shown that children who are younger and less advanced in terms of speech development exhibit better articulation and resonance than those who have their palates repaired when they are older and exhibit more speech and language development (Chapman, Hardin-Jones, Goldstein, et al, 2008). Persistent velopharyngeal insufficiency, manifested by nasal regurgitation, audible nasal emission, and hypernasal speech, may require a secondary surgical procedure. Nasendoscopy and/or videofluoroscopy may be used to determine whether further surgery is needed to enhance the function of the palate. Once secondary surgical procedures for velopharyngeal insufficiency have been completed, children exhibiting speech production errors will often require intensive speech therapy to correct acquired patterns. Alveolar bone grafting may be needed to place bone in the area of the alveolar cleft, to allow structure for anchoring of the secondary teeth during eruption.
Long-Term Problems: The care of children with CL and CP often involves a team of specialists who meet periodically to examine the child and consult with one another and with the parents. Even with adequate anatomic closure, many children with CL and CP have some degree of speech impairment that requires speech therapy. Speech production errors in children with clefts are often related to inefficient function of the muscles of the soft palate and nasopharynx (which lead to the development of compensatory speech patterns), improper tooth alignment, and varying degrees of hearing loss.
Improper drainage of the middle ear, as a result of inefficient function of the eustachian tube secondary to the CP, causes increased pressure in the middle ear and contributes to recurrent middle ear effusion or otitis media, which can lead to hearing impairment in some children with CP. The insertion of pressure-equalizing tubes has become standard procedure in the child with CL/CP and is often performed at the same time as other surgical procedures (such as CL repair) to facilitate fluid drainage from the middle ear and prevent middle ear effusion and recurrent otitis media.
Extensive orthodontics and prosthodontics are usually needed to correct malposition of the teeth and maxillary arches. Teeth may be missing, malformed, or malpositioned, which can interfere with speech and feeding. In addition, a significant number of these children have an inadequate nasal airway that forces them to breathe through the mouth, which also contributes to oral deformity. Children with both CL and CP often require several stages of orthodontic therapy. An orthopedic appliance is often worn 24/7 starting in the first week of life to align the maxillary segments into a near-normal relationship up until CL repair at 3 to 5 months. This is frequently done to facilitate a primary lip closure. The second stage (at 2 to 5 years) consists of palatal expansion and correction of a dental crossbite (a condition in which the upper teeth close inside the lower teeth) in an attempt to allow the primary teeth to develop in a normal relationship. The third stage of therapy (at 10 to 11 years) takes place during the mixed-dentition stage and involves correction of faulty occlusion. In the fourth stage (at 12 to 18 years) treatment of the permanent teeth is accomplished in much the same manner as for any adolescent except for alignment and spacing in the cleft area.
Often temporary or permanent dental prostheses are necessary to replace missing teeth. These assist in chewing and produce a more pleasing cosmetic effect. Special dental plates, called obturators, are sometimes used to mechanically close clefts in the palate to facilitate feeding and speech until permanent closure is attempted. Any appliance must be checked periodically to ensure a proper fit and see that it is performing its intended function.
A major problem for a child with CP may be compensatory speech pattern. This can occur as a result of any or all of the previously discussed complications: insufficient palate function, abnormal dentition, and hearing loss. CP interferes with speech sounds in the mouth that are normally made through interaction of the velar and pharyngeal muscles. Children with CP may develop compensatory patterns if velopharyngeal closure is compromised. These errors can be very difficult to correct, so early intervention focusing on prevention of speech errors is strongly advocated. Improper tooth alignment can pose a mechanical hazard to development of clear speech, and hearing loss from middle ear infection or middle ear effusion is an additional impediment because of difficulty in interpreting sounds. With isolated CL, no speech problems should be anticipated. The child with CP usually requires the services of a speech-language pathologist.
Nursing Care Management
The immediate nursing problems in the care of an infant with CL and CP deformities are related to feeding the infant and dealing with the parental reaction to the defect. Facial deformities are particularly disturbing to parents. CL, especially, is a disfiguring, visible defect that may generate a strong negative response in parents. During the initial phase after birth of an infant with CL or CP, it is important for the nurse to address not only the infant’s physical needs but also the parents’ emotional needs.
The nurse should encourage expression of parental grief and fears. Such expression may promote attachment in the preoperative period. It is especially important to emphasize the positive aspects of the infant’s physical appearance and to express optimism regarding surgical correction while acknowledging the parents’ concern. The manner of handling the infant should convey to the parents that the infant is indeed a precious human being. (See Birth of a Child with a Physical Defect, p. 391.)
Feeding: Feeding the newborn with CL/CP can be a challenge, and teaching the parent to successfully feed the child is perhaps one of the most significant and challenging nursing roles. Growth failure in these infants has been attributed to preoperative feeding difficulties; however, such difficulties can be overcome by increasing overall caloric intake with a higher-calorie formula, nutrition counseling, and evaluation. Weekly weight checks at the practitioner’s office assist in monitoring the infant’s weight preoperatively. After surgical repair most infants with isolated CL or CP and no associated syndrome gain weight successfully and achieve adequate weight and height for age.
Although some infants with isolated CL are typically able to breast-feed without difficulty, breast-feeding is a difficult process for most infants with an unrepaired CP. La Leche League International reports that “over time, lactation consultants have found that feeding exclusively at the breast is a difficult goal for all but a few infants with uncorrected cleft palates” (Cleft Palate Foundation, 2008). An infant with CP is unable to achieve adequate suction to extract breast milk directly from the breast. Clefts of the palate reduce the infant’s ability to suck, which interferes with compression of the areola and renders breast-feeding and traditional bottle-feeding difficult. Standard bottle nipples may be unsuitable for these infants, who are unable to generate the suction required. Therefore special nipples or other feeding devices are needed. Liquid taken into the mouth tends to escape via the CP through the nose. Accepting that she may not be able to breast-feed can be difficult for a new mother. However, the Cleft Palate Foundation recommends that a mother of a child with a cleft should be encouraged to try the following strategies:
• She may try pumping her breast milk and providing the milk with an adapted bottle made for children with clefts.
• Skin-to-skin contact should be encouraged when possible.
• After bottle-feeding, the infant may be put to the mother’s breast for nonnutritive sucking.
With appropriate adaptive bottles and positioning, infants with clefts can be efficient oral feeders. Feeding is best accomplished with the infant’s head in an upright position, either held in the caregiver’s hand or cradled in the arm.
A number of special feeding devices are available for feeding the infant with CL/CP, and some are more successful than others, depending on a number of factors (Fig. 11-18). One device is the Cleft Lip/Cleft Palate Nurser, which consists of a squeezable plastic bottle and a cross-cut nipple. The Haberman Special Needs Feeder may also be used successfully in infants with a poor or disorganized suck. The Haberman feeder has a specially designed valve and nipple to adjust the flow of milk to the infant and prevent choking or gagging. The nipple chamber of the Haberman bottle is large, which allows the feeder to provide extra assistance by squeezing the chamber if needed. The Pigeon bottle has a nipple with a Y-cut and is slightly larger and more bulbous to fit naturally into the oral cavity. A one-way back-flow valve prevents milk from flowing retrograde into the bottle to minimize the amount of air the infant swallows. The Pigeon bottle is not a squeezable feeding system.
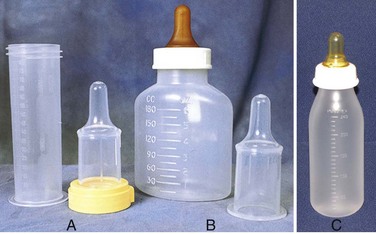
Fig. 11-18 A, Haberman feeder. B, Mead Johnson bottle used to feed an infant with cleft lip and palate. C, Pigeon bottle. (A and B, Courtesy Texas Children’s Hospital, Houston. C, Courtesy Paul Vincent Kuntz, Texas Children’s Hospital, Houston.)
Using these various types of nipples for feeding also has the advantage of helping to meet the infant’s sucking needs. Muscle development is especially important for later development of speech. The Haberman and the Pigeon nipples work by compression, so the nipple is positioned in such a way that it is compressed by the infant’s tongue and existing palate. If a single-slit nipple is used (such as the Haberman feeder), the slit is placed vertically so that the infant will be able to produce and stop the flow of milk by alternately opening and closing the opening. Regardless of the type of nipple used, the person doing the feeding should resist the temptation to remove the nipple because of the noise the infant makes or because of fear that the infant will choke. An indication that the infant needs to stop feeding momentarily is the facial signal, which involves elevated eyebrows and a wrinkled forehead; the nipple may be gently removed to allow the infant to swallow formula in the mouth without getting upset. These infants need frequent burping because they have a tendency to swallow excessive amounts of air.
Some CP specialists advocate for the use of feeding obturators to assist with feeding. However, these devices do not improve feeding efficiency or growth within the first year of life (Masarei, Wade, Mars, et al, 2007).
Regardless of the feeding method used, the mother should begin to feed the infant as soon as possible. In this way she is able to help determine the method best suited to her and the infant and to become adept in the technique before discharge.
Preoperative Care: In preparation for the surgical repair, instruct the parents to accustom the infant to some of the needs of the early postoperative period. Some craniofacial surgery teams encourage the transition to cup-feeding before CP surgery, and this feeding method is used postoperatively as well. Preoperative preparation, including medication, is determined by the surgeon and anesthesiologist.
Postoperative Care: Cleft Lip: The major efforts in the postoperative period are directed toward protecting the operative site. Avoid the prone position to prevent suture damage. After CL repair (cheiloplasty), some surgeons allow breast-feeding or syringe feeding once the child is awake and alert. Cheiloplasty is often performed on a day-of-surgery basis with discharge to home after ascertainment of adequate fluid intake.
Elbow restraints may be applied immediately after surgery to prevent the infant from rubbing the suture line, although many surgeons no longer support this practice.
Pain management should continue in the home setting, and parents are taught how to administer the appropriate dosage of analgesic. Clear liquids are offered when the infant has fully recovered from the anesthesia, and breast- or syringe-feeding is usually resumed as tolerated. A thin layer of antibiotic ointment may be prescribed for application to the suture line. Meticulous care of the suture line is a nursing responsibility. Inflammation or infection interferes with optimum healing and the ultimate cosmetic effect of the surgical repair.
 NURSING ALERT
NURSING ALERT
Avoid the use of suction or objects in the mouth such as tongue depressors, thermometers, spoons, or straws.
The infant should be positioned to prevent airway obstruction by secretions, blood, or the tongue. Gentle aspiration of mouth and nasopharyngeal secretions may be necessary to prevent aspiration and respiratory complications. Because of vascularity of the lip and palate, postoperative care involves monitoring operative sites for bleeding. Excessive swallowing may be a sign of bleeding and swallowing blood.
Postoperative Care: Cleft Palate: The child with a CP repair (palatoplasty) can lie on the abdomen, especially immediately postoperatively. The child may resume feedings by special feeding device shortly after surgery. Acceptable feeding devices include soft-tip (like a nipple) sippy cups, an open cup, an oropharyngeal syringe, and specialized bottles with soft tubing.
The speech-language pathologist evaluates the child’s individual needs and directs the parents in specific activities to facilitate speech development. The more children use speech, the sooner they will gain self-confidence and assurance in social situations.
Throughout the child’s therapy, the ultimate goal should be the development of a healthy personality and self-esteem. Several agencies provide services and information for children with CL/CP and their families. These include the Cleft Palate Foundation,* Birth Defect Research for Children, Inc.,† and the March of Dimes.‡
Sometimes the infant has difficulty breathing after surgery because of swelling. Humidified air may be provided as blow-by to alleviate edema of the tissues, and the side-lying or semireclined positioning may be used to facilitate drainage of secretions.
Elbow restraints can keep the hands away from the mouth, and the parents should maintain this precaution at home until the palate is healed. When used the restraints may be removed with adequate supervision to allow the child to exercise the arms. Assess the infant for pain postoperatively. Opiates may be prescribed for the first 24 to 48 hours, or longer if needed, and acetaminophen may be given thereafter (see Nursing Care Plan).
 NURSING CARE PLAN
NURSING CARE PLAN
The older infant is usually discharged on a blenderized or soft diet, and parents should continue the diet until the surgeon directs them to do otherwise. Caution them against allowing the child to eat hard items such as toast, hard cookies, or potato chips, which could damage the newly repaired palate.
Preparation for Discharge and Home Care: Parents should participate in the infant’s care as soon as possible after surgery. They should learn the proper feeding method. Parents should also know how to cleanse the suture line to free any crust that might form and to replace elbow restraints (if used). Carefully evaluate and discuss car seat restraint appropriate to the infant’s condition with the parents before discharge. Also discuss the infant sleep position based on the infant’s condition and the American Academy of Pediatrics recommendation for supine sleeping in infants. (See Chapter 13.)
Long-Term Family Guidance: The problems of parents in the care of an infant with CP may extend well beyond the initial acceptance of and adjustment to the defect and surgical correction. These families need support and encouragement by health care professionals and guidance in activities that facilitate the most normal life for the child.
Parents often cite financial stressors as being the most difficult issue to deal with when the child has a craniofacial anomaly. However, with the combined efforts of the family and the health care team, the majority of these infants achieve a satisfactory long-term outcome.
Parents need to understand the therapy and the purpose of any appliance. They should learn proper care and placement of the device and that establishing good mouth care and proper brushing habits is especially important for these children.
Because of the increased risk of middle ear infection, the ears are examined regularly, and hearing tests are scheduled early and repeated periodically throughout childhood. It is important to emphasize the need for an ear examination when the child has symptoms of an upper respiratory tract infection. When treatment can be implemented early, the chances are greater that permanent damage to the ear can be avoided. Parents should be alert for signs of any hearing impairment in the child so they can obtain needed help and prevent progression of any deficit. (See Chapter 24.)
The parents also need guidance in helping the child develop normal speech. They should encourage the child’s early attempts to make sounds. Some parents erroneously believe that the child may form poor speech habits if he or she tries to speak before the palate is repaired. Before palatal repair children should make the nasal consonants “m,” “n,” and “ng” (as “ing”), as well as “w” and “y” and most vowels. After palatal surgery children should attempt to produce most consonants, although speech therapy may be required to facilitate production of these sounds (see Nursing Care Plan).
Esophageal Atresia and Tracheoesophageal Fistula
Congenital esophageal atresia (EA) and tracheoesophageal fistula (TEF) are rare malformations that represent a failure of the esophagus to develop as a continuous passage and a failure of the trachea and esophagus to separate into distinct structures. These defects may occur as separate entities or in combination, and without early diagnosis and treatment they pose a serious threat to the infant’s well-being.
The incidence is estimated to be approximately 2 in 10,000 live births (Achildi and Grewal, 2007). There appears to be a slightly higher incidence in males, and the birth weight of most affected infants is significantly lower than average, with an unusually high incidence of preterm birth in infants with EA and a subsequent increase in mortality. A history of maternal polyhydramnios is common.
Approximately 50% of the cases of EA/TEF are a component of VATER or VACTERL association, acronyms used to describe associated anomalies (VATER for Vertebral defects, imperforate Anus, Tracheoesophageal fistula, and Radial and Renal dysplasia; and VACTERL for Vertebral, Anal, Cardiac, Tracheal, Esophageal, Renal, and Limb) (Orenstein, Peters, Khan, et al, 2007). Cardiac anomalies may also occur with EA/TEF; therefore all patients should undergo a workup for associated anomalies.
Pathophysiology
The esophagus develops from the first segment of the embryonic gut. During the fourth and fifth weeks of gestation, the foregut normally lengthens and separates longitudinally. Each longitudinal portion fuses to form two parallel channels (the esophagus and the trachea) that are joined only at the larynx. Anomalies involving the trachea and esophagus are caused by defective separation, incomplete fusion of the tracheal folds after this separation, or altered cellular growth during embryonic development.
The most commonly encountered form of EA and TEF (80% to 90% of cases) is one in which the proximal esophageal segment terminates in a blind pouch and the distal segment is connected to the trachea or primary bronchus by a short fistula at or near the tracheal bifurcation (Fig. 11-19, C). The second most common type (7% to 8%), or “pure EA,” consists of a blind pouch at each end, widely separated and with no communication to the trachea (Fig. 11-19, A). An H-type EA refers to an otherwise normal trachea and esophagus connected by a fistula (4% to 5%) (Fig. 11-19, E). Extremely rare anomalies involve a fistula from the trachea to the upper esophageal segment (0.8%) (Fig. 11-19, B) or to both the upper and lower segments (0.7% to 6%) (Fig. 11-19, D).
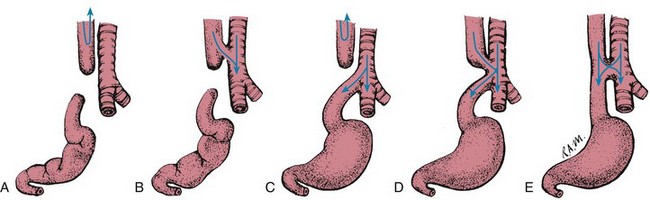
Clinical Manifestations
The presence of EA is suspected in a newborn with frothy saliva in the mouth and nose, drooling, choking, and coughing. Respiratory distress may be mild or significant, depending on the type of defect and the infant’s gestational age. If fed, the infant may swallow normally but suddenly cough and gag, with return of fluid through the nose and mouth. The infant may become cyanotic and apneic because of aspiration of formula or saliva.
In the infant who has EA with a distal TEF (type C), the stomach becomes distended with air, and thoracic and abdominal compression (especially during crying) cause the gastric contents to be regurgitated through the fistula and into the trachea, producing a chemical pneumonitis. When the upper segment of the esophagus opens directly into the trachea (types B and D), the infant is in danger of aspirating any swallowed material. Cyanosis or choking during feeding may be the only symptom of type E fistula (see Fig. 11-19). The child with this H type of EA may not manifest symptoms until later in life when he or she shows signs of chronic respiratory problems, recurrent pneumonia, and signs of gastroesophageal reflux (Orenstein, Peters, Khan, et al, 2007).
Diagnostic Evaluation
In a newborn who has symptoms suggestive of EA/TEF, an attempt is made to pass an NG-orogastric (OG) catheter into the esophagus. Inability to pass the catheter warrants further evaluation.
Although the diagnosis is established on the basis of clinical signs and symptoms, the exact type of anomaly is determined by radiographic studies. A radiopaque catheter is inserted into the hypopharynx and advanced until it encounters an obstruction. Chest films are taken to ascertain esophageal patency or the presence and level of a blind pouch. Films that show air in the stomach indicate a connection between the trachea and the distal esophagus in types C, D, and E. Complete absence of air in the stomach is seen in types A and B. Occasionally, fistulas are not patent, which makes their presence more difficult to diagnose. A careful bronchoscopic examination may be performed in an attempt to visualize the fistula.
The presence of polyhydramnios (accumulation of >2000 ml of amniotic fluid) prenatally is a clue to the possibility of EA in the unborn infant, especially with defect type A, B, or C. With these types of EA/TEF, amniotic fluid normally swallowed by the fetus is unable to reach the GI tract to be absorbed and excreted by the kidneys. The result is an abnormal accumulation of amniotic fluid, or polyhydramnios.
Therapeutic Management
The treatment of EA and TEF includes maintenance of a patent airway, prevention of pneumonia, gastric or blind pouch decompression, supportive therapy, and surgical repair of the anomaly.
When EA with a TEF is suspected, the infant is immediately deprived of oral intake, IV fluids are initiated, and the infant is positioned to facilitate drainage of secretions and decrease the likelihood of aspiration. Accumulated secretions are suctioned frequently from the mouth and pharynx. A double-lumen catheter should be placed into the upper esophageal pouch and attached to intermittent or continuous low suction. The infant’s head is kept upright to facilitate removal of fluid collected in the pouch and to prevent aspiration of gastric contents. Broad-spectrum antibiotic therapy is often instituted if there is a concern about aspiration of gastric contents.
Surgical Correction: Most malformations can be corrected surgically in one operation or in two or more staged procedures. The success depends on early diagnosis before complications occur and on the presence and severity of associated anomalies and illness factors, including preterm birth. With measures instituted to prevent aspiration pneumonia and to ensure adequate hydration and nutrition, surgery may be postponed to allow for more effective treatment of pneumonia and physiologic stabilization so that the infant can better withstand the complex surgery. The delay also offers an opportunity for further evaluation and assessment to rule out any associated anomalies and to optimize respiratory support.
The surgery consists of a thoracotomy with division and ligation of the TEF and an end-to-end or end-to-side anastomosis of the esophagus. A chest tube may be inserted to drain intrapleural air and fluid. For infants who are not stable enough to undergo definitive repair or those with a lengthy gap between the proximal and distal esophagus, a staged operation is preferred that involves gastrostomy, ligation of the TEF, and constant drainage of the esophageal pouch. A delayed esophageal anastomosis is usually attempted after several weeks to months. In some centers thoracoscopic repair of EA/TEF is being used successfully, thus negating the need for a thoracotomy and minimizing associated postoperative complications and morbidities (Achildi and Grewal, 2007; Holcomb, Rothenberg, Bax, et al, 2005).
Further surgical techniques may be performed later to facilitate esophageal lengthening. If an esophageal anastomosis cannot be accomplished, a cervical esophagostomy (to allow drainage of saliva through a stoma in the neck) and gastrostomy are performed.
A primary anastomosis may be impossible because of insufficient length of the two segments of esophagus. This occurs if the distance between the two segments is 3 to 4 cm (1.2 to 1.6 inches) or greater; this is often referred to as long-gap EA (Orenstein, Peters, Khan, et al, 2007). In these cases an esophageal replacement procedure using a part of the colon or gastric tube interposition may be necessary to bridge the missing esophageal segment.
Tracheomalacia may occur as a result of weakness in the tracheal wall that exists when a dilated proximal pouch compresses the trachea early in fetal life. It may also occur as a result of inadequate intratracheal pressure causing abnormal tracheal development. Clinical signs of tracheomalacia include barking cough, stridor, wheezing, recurrent respiratory tract infections, cyanosis, and, sometimes, apnea. Tracheomalacia may occur in up to as many as 75% of children with EA/TEF but may be clinically significant in only 10% to 20% of infants with EA/TEF; surgical intervention is required in severe cases (Achildi and Grewal, 2007).
Prognosis: The survival rate is nearly 100% in otherwise healthy children. Most deaths are the result of extreme prematurity or other lethal associated anomalies.
Potential complications after the surgical repair of EA and TEF depend on the type of defect and surgical correction. Complications of repair include an anastomotic leak, strictures caused by tension or ischemia, esophageal motility disorders causing dysphagia, respiratory compromise, and gastroesophageal reflux. Anastomotic esophageal strictures may cause dysphagia, choking, and respiratory distress. The strictures are often treated with routine esophageal dilation. Feeding difficulties are often present for months or years postoperatively, and the infant must be monitored closely to ensure adequate weight gain, growth, and development. In some cases laparoscopic fundoplication may be required. At times the infant must be fed via gastrostomy or jejunostomy to provide adequate caloric intake.
Nursing Care Management
The nursing process in the care of the infant with EA/TEF is described in the Nursing Care Plan on pp. 438-439.
Nursing responsibility for detection of this serious malformation begins immediately after birth. For the infant with the classic signs and symptoms of EA (see Clinical Manifestations, p. 435) the major concern is the establishment of a patent airway and prevention of further respiratory compromise. Cyanosis is usually a result of laryngeal spasm caused by overflow of saliva into the larynx from the proximal esophageal pouch or aspiration; it normally resolves after removal of the secretions from the oropharynx by suctioning. The passage of a small-gauge OG feeding tube via the mouth into the stomach during the initial nursing physical assessment is helpful to rule out EA or other obstructive defects. Additional stabilization and nursing care are discussed in the next section.
NURSING ALERT
Any infant who has an excessive amount of frothy saliva in the mouth or difficulty with secretions and unexplained episodes of apnea, cyanosis, or oxygen desaturation should be suspected of having an EA/TEF and referred immediately for medical evaluation.
Preoperative Care: The nurse carefully suctions the mouth and nasopharynx and places the infant in an optimum position to facilitate drainage and avoid aspiration. The most desirable position for a newborn who is suspected of having the typical EA with a TEF (e.g., type C) is supine (or sometimes prone) with the head elevated on an inclined plane of at least 30 degrees. This positioning minimizes the reflux of gastric secretions at the distal esophagus into the trachea and bronchi, especially when intraabdominal pressure is elevated.
It is imperative to immediately remove any secretions that can be aspirated. Until surgery the blind pouch is kept empty by intermittent or continuous suction through an indwelling double-lumen or Replogle catheter passed orally or nasally to the end of the pouch. In some cases a percutaneous gastrostomy tube is inserted and left open so that any air entering the stomach through the fistula can escape, thus minimizing the danger of gastric contents being regurgitated into the trachea. The gastrostomy tube is emptied by gravity drainage. Feedings through the gastrostomy tube and irrigations with fluid are contraindicated before surgery in the infant with a distal TEF.
Nursing interventions include respiratory assessment, airway management, thermoregulation, fluid and electrolyte management, and parenteral nutritional support.
Often the infant must be transferred to a hospital with a specialized care unit and pediatric surgical team. The nurse advises the parents of the infant’s condition and provides them with necessary support and information.
Postoperative Care: Postoperative care for these infants is the same as for any high-risk newborn. Adequate thermoregulation is provided, the double-lumen NG catheter is attached to low-suction or gravity drainage, parenteral nutrition is provided, and the gastrostomy tube (if applicable) is returned to gravity drainage until feedings are tolerated. If a thoracotomy is performed and a chest tube is inserted, attention to the appropriate function of the closed drainage system is imperative. Pain management in the postoperative period is important even if only a thoracoscopic approach is used. In the first 24 to 36 hours the nurse should provide pain management for the neonate just as for an adult undergoing a similar procedure. (See Neonatal Pain, Chapter 7.) Tracheal suction should only be done using a premeasured catheter and with extreme caution to avoid injury to the suture line.
If tolerated, gastrostomy feedings may be initiated and continued until the esophageal anastomosis is healed. Before oral feedings are initiated and the chest tube (if applicable) is removed, a contrast study or esophagram will verify the integrity of the esophageal anastomosis.
The nurse must carefully observe the initial attempt at oral feeding to make certain the infant is able to swallow without choking. Oral feedings are begun with sterile water, followed by frequent small feedings of breast milk or formula. Until the infant is able to take a sufficient amount by mouth, oral intake may need to be supplemented by bolus or continuous gastrostomy feedings. Ordinarily infants are not discharged until they can take oral fluids well. The gastrostomy tube may be removed before discharge or maintained for supplemental feedings at home.
Special Problems: Upper respiratory tract complications are a threat to life in both the preoperative and postoperative periods. In addition to pneumonia, the infants are in constant danger of respiratory distress resulting from atelectasis, pneumothorax, and laryngeal edema. Report any persistent respiratory difficulty after removal of secretions to the surgeon immediately. Monitor the infant for anastomotic leaks and signs of infection such as purulent chest tube drainage, an increased white blood cell count, and temperature instability.
In the infant awaiting esophageal replacement surgery, the upper esophageal segment may be drained by means of a cervical esophagostomy. An esophagostomy is difficult to care for because the skin may become irritated by moisture from the continuous discharge of saliva. Frequent removal of drainage followed by application of a layer of protective ointment, barrier dressing, and/or a collection device is usually sufficient treatment. An enterostomal nurse may provide helpful guidance in the prevention and treatment of skin breakdown.
For the infant who requires esophageal replacement, provide nonnutritive sucking with a pacifier. Sometimes food and fluid are given orally (sham feedings); the intake drains from the esophagostomy but allows the infant to develop mature sucking patterns and, with other appropriate oral stimulation, can prevent feeding resistance. (See Chapter 10.) Infants who take nothing by mouth (NPO) for an extended period and have not received oral stimulation frequently have difficulty eating by mouth after corrective surgery and may develop oral hypersensitivity and feeding aversion. They require patient, firm guidance in learning the techniques of taking food into the mouth and swallowing after repair. A referral to a multidisciplinary feeding behavior team may be necessary.
Family Support, Discharge Planning, and Home Care: One of the difficulties in TEF is the immediate transfer of the sick infant to the intensive care unit and sometimes lengthy hospitalization. Parent-infant bonding is facilitated by encouraging parents to visit the infant, participate in his or her care when appropriate, and express their feelings regarding the infant’s condition. The nurse in the intensive care unit should assume responsibility for ensuring that the parents are fully informed of the infant’s progress.
Preparing parents for discharge of their infant involves teaching the techniques that will be continued at home. The parents also learn signs of respiratory difficulty and of esophageal stricture (poor feeding, dysphagia, drooling, regurgitating undigested food) and gastroesophageal reflux.
Parents must be aware of dietary restrictions. Remind parents that it is particularly important to guard against the infant swallowing foreign objects. They should cut solid food into small pieces, teach the child to chew thoroughly, give frequent sips of liquid to help swallow food, and avoid foods such as whole hot dogs or large pieces of meat that may become lodged in the esophagus. (See Injury Prevention, Chapter 12.)
Many of these infants have some degree of tracheomalacia; therefore parents should be educated regarding the signs and symptoms. Discharge planning should include teaching parents and other caretakers infant cardiopulmonary resuscitation. Because many infants with EA and a TEF develop gastroesophageal reflux, precautions should be initiated. (See Gastroesophageal Reflux, Chapter 33.)
Discharge planning should include attainment of needed equipment and home nursing services to assist with ongoing assessment of the child and continuity of care (see Nursing Care Plan). (See Chapter 25.)

Anorectal Malformations
Anorectal malformations are among the more common congenital malformations caused by abnormal development, with an incidence of approximately 1 in 5000 births (Levitt and Peña, 2007). These malformations may range from simple imperforate anus to include other associated complex anomalies of GU and pelvic organs, which may require extensive treatment for fecal, urinary, and sexual function. Anorectal malformations may occur in isolation or as a part of the VACTERL association (see p. 435). These anomalies are classified according to the newborn’s gender and abnormal anatomic features, including GU defects (Box 11-7).
Rectal atresia and stenosis occur when the anal opening appears normal, there is a midline intergluteal groove, and usually no fistula exists between the rectum and urinary tract. Rectal atresia is a complete obstruction (inability to pass stool) and requires immediate surgical intervention. Rectal stenosis may not become apparent until later in infancy when the infant has a history of difficult stooling, abdominal distention, and ribbonlike stools.
A persistent cloaca is a complex anorectal malformation in which the rectum, vagina, and urethra drain into a common channel opening into the perineum (Fig. 11-20, A).
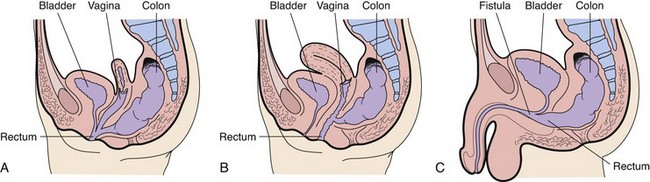
Fig. 11-20 Anorectal malformations. A, Typical cloaca (female). B, Low rectovaginal fistula (female). C, Rectourethral bulbar fistula (male).
Imperforate anus includes several forms of malformation without an obvious opening (Fig. 11-21). Frequently a fistula (an abnormal communication) leads from the distal rectum to the perineum or GU system (Fig. 11-20, B and C). The fistula may be evidenced when meconium is evacuated through the vaginal opening, the perineum below the vagina, the male urethra, or the perineum under the scrotum. The presence of meconium on the perineum does not indicate anal patency. A fistula may not be apparent at birth, but as peristalsis increases, meconium is forced through the fistula into the urethra or onto the newborn’s perineum.
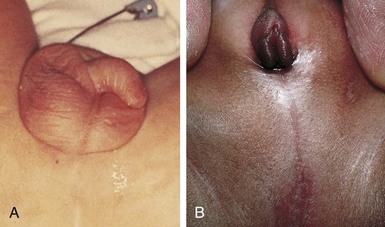
Fig. 11-21 A, No visible external opening is consistent with high imperforate anus defect; absence of intergluteal cleft is also common. B, Imperforate anus in female, commonly associated with cloacal anomaly, which manifests as a single perineal opening on perineum. (From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 5, St Louis, 2007, Mosby.)
Pathophysiology
During embryonic development the cloaca becomes the common channel for the developing urinary, genital, and rectal systems. The cloaca is divided at the sixth week of gestation into an anterior urogenital sinus and a posterior intestinal channel by the urorectal septum. After the lateral folds join the urorectal septum, separation of the urinary and rectal segments takes place. Further differentiation results in the anterior GU system and the posterior anorectal channel. An interruption of this development leads to incomplete migration of the rectum to its normal perineal position.
Diagnostic Evaluation
The diagnosis of an anorectal malformation is based on the physical finding of an absent anal opening. Other symptoms may include abdominal distention, vomiting, absence of meconium passage, or presence of meconium in the urine. Additional physical findings with an anorectal malformation are a flat perineum and the absence of a midline intergluteal groove. The appearance of the perineum alone does not accurately predict the extent of the defect and associated anomalies. GU and spinal-vertebral anomalies associated with anorectal malformations should be considered when an anomaly is noted. EA with or without TEF, cardiac defects, and spinal or vertebral anomalies may occur in association with anorectal malformations, and the infant should be carefully evaluated for the presence of these and other anomalies.
A perineal fistula (see Box 11-7) may be diagnosed by clinical observation. The presence of a prominent anal dimple and a band of skin tissue commonly known as a bucket handle is indicative of a perineal fistula (Levitt and Peña, 2007). Abdominal and pelvic ultrasonography is performed to further evaluate the infant’s anatomic malformation. An IV pyelogram and a voiding cystourethrogram (VCUG) are performed to evaluate associated anomalies involving the urinary tract. Other diagnostic examinations that may be performed include a pelvic MRI, radiography, ultrasound, and fluoroscopic examination of pelvic anatomic contents and lower spinal anatomy.
Therapeutic Management
The primary management of anorectal malformations is surgical. Once the defect is identified, take steps to rule out associated life-threatening defects, which need immediate surgical intervention. Provided no immediate life-threatening problems exist, the newborn is stabilized and kept NPO for further evaluation. IV fluids are provided to maintain glucose and fluid and electrolyte balance. Current recommendation is that surgery be delayed at least 24 hours to properly evaluate for the presence of a fistula and possibly other anomalies (Levitt and Peña, 2007).
The surgical treatment of anorectal malformations varies according to the defect but usually involves one, or possibly a combination of several, of the following procedures: anoplasty, colostomy, posterior sagittal anorectoplasty (PSARP) or other pull-through with colostomy, and colostomy (take-down) closure. The Nursing Care Management discussion below outlines some aspects of preoperative and postoperative care.
A primary laparoscopic repair (without colostomy) of some anorectal malformations is being performed successfully in some centers. This minimizes surgical risks, associated morbidity, and postoperative pain management.
Prognosis: The long-term prognosis depends on such factors as the type of defect, anatomy of the sacrum and vertebrae, quality of muscles, and the success of the surgery. Levitt and Peña (2005) emphasize that each defect has a different prognosis and that the prognosis varies according to individual presentation.
The presence of a flat or “rocker” bottom and no midline groove usually carries a poor prognosis for bowel continence because of associated neurologic, muscular, and anatomic problems. When the internal anal sphincter is absent, incontinence is a common long-term problem. These children may achieve socially acceptable continence over time with the aid of a bowel management program. Other potential complications after surgical treatment of anorectal anomalies include strictures, recurrent rectourinary fistula, mucosal prolapse, and constipation.
Nursing Care Management
The first nursing responsibility is assisting in identification of anorectal malformations. A newborn who does not pass stool within 24 hours after birth or has meconium that appears at a location other than the anal opening requires further assessment. Preoperative care includes diagnostic evaluation, GI decompression, bowel preparation, and IV fluids.
For the newborn with a perineal fistula, an anoplasty is performed, which involves moving the fistula opening to the center of the sphincter and enlarging the rectal opening. Postoperative nursing care after anoplasty is primarily directed toward healing the surgical site without other complications. A program of anal dilations is usually initiated when the child returns for the 2-week check-up. Feedings are started soon after surgical repair, and breast-feeding is encouraged because it causes less constipation.
In neonates with anomalies such as cloaca (female), rectourethral prostatic fistula (male), and vestibular fistula (female), a descending colostomy is performed to allow fecal elimination and avoid fecal contamination of the distal imperforate section and subsequent urinary tract infection in infants with urorectal fistulas. With a colostomy, postoperative nursing care is directed toward maintaining appropriate skin care at the stoma sites (both distal and proximal), managing postoperative pain, and administering IV fluids and antibiotics. Postoperative NG decompression may be required with laparotomy, and nursing care focuses on maintenance of appropriate drainage. (See Chapter 27 for colostomy care.)
The PSARP is a common surgical procedure for the repair of anorectal malformations in infants approximately 1 to 2 months after the initial colostomy. Preoperative PSARP care often involves irrigation of the distal stoma to prevent fecal contamination of the operative site. During this time parents must be given accurate yet simple information regarding the infant’s appearance postoperatively and expectations as to their level of involvement in the child’s care.
In the PSARP procedure the repair is made via a posterior midline sacral approach to dissect the different muscle groups involved without damaging strategic innervation of pelvic structures, so that optimum postoperative bowel continence is achieved. A laparotomy may be required if the rectum is unidentifiable by the posterior approach. Additional management after successful repair involves a program of anal dilations, colostomy closure, and a bowel management program.
Parents are instructed in perineal and wound care or care of the colostomy as needed. Anal dilations may be necessary for some infants. Parents should observe stooling patterns and observe for signs of anal stricture or complications. Information on dietary modifications and administration of medications is included in counseling. Nurses have a vital role in helping families of a child with anorectal malformations provide optimum care so that bowel management is successful and quality of life enhanced for the child and family.
Family Support, Discharge Planning, and Home Care: Long-term follow-up care is essential for children with complex malformations. Parents need reassurance when a colostomy is performed regarding the child’s appearance and their ability to care for the child at home. With much patience and reassurance, parents learn how to provide optimum care of the skin and the appliance, while maintaining an appropriate bond with the child.
After the definitive pull-through procedure, toilet training may be delayed. Complete continence is seldom achieved at the usual age of 2 to 3 years. Bowel habit training, bowel management irrigation programs, diet modification, and administration of stool softeners or fiber help children improve bowel function and social continence. Some children never achieve bowel continence and must rely on daily bowel irrigations. Support and reassurance during the slow progression to normal, socially acceptable function are essential.
Biliary Atresia
Biliary atresia, or extrahepatic biliary atresia (EHBA), is a progressive inflammatory process that causes both intrahepatic and extrahepatic bile duct fibrosis, resulting in eventual ductal obstruction. The incidence of biliary atresia is approximately 1 in 10,000 to 15,000 live births (A-Kader and Balistreri, 2007; Kelly and Davenport, 2007). Associated malformations include polysplenia, intestinal atresia, and malrotation of the intestine. Biliary atresia, if untreated, usually leads to cirrhosis, liver failure, and death in the first 2 years of life.
Pathophysiology
The exact cause of biliary atresia is unknown, although immune mechanisms or viral injury may be responsible for the progressive process that results in complete obliteration of the bile ducts. Biliary atresia is not seen in the fetus or stillborn or newborn infant. This suggests that biliary atresia is acquired late in gestation or in the perinatal period and is manifested a few weeks after birth. Congenital infections such as cytomegalovirus, rubella virus, Epstein-Barr virus, rotavirus, and reovirus type 3 have been implicated as a cause of hepatocellular damage leading to biliary atresia, yet no specific agent is identified in every case. Immune-mediated bile duct injury from viral exposure and immaturity of the neonatal immune system may play a role in the destruction of bile ducts and development of EHBA. Other potential causes include an early first trimester insult to the developing bile ducts or a postnatal viral insult; genetic factors may also play a role in the pathogenesis (Davenport, 2005). Early in the course of the disease, the intrahepatic ducts are patent from the interlobular ductules to the porta hepatis. The size of these structures is variable and is correlated with the infant’s age and with bile excretion after surgical treatment. These structures are present in most affected infants under 2 months of age but gradually disappear over the next few months and by 4 months are completely replaced by fibrous tissue.
The degree of involvement of the extrahepatic biliary ducts is also variable. Most commonly the entire extrahepatic system is involved in the obliterative process, but some infants have a patent proximal portion of the extrahepatic duct or patency of the gallbladder, cystic duct, and common bile duct. Microscopic examination of the liver tissue reveals cholestasis with absent or diminished bile duct proliferation and fibrosis.
Clinical Manifestations
Many infants with biliary atresia are full term and appear healthy at birth. If jaundice persists beyond 2 weeks of age, especially if the direct (conjugated) serum bilirubin is elevated, the nurse should suspect biliary atresia. The urine may be dark, and the stools often become progressively acholic or gray, indicating absence of bile pigment. Hepatomegaly is present early in the course of the disease, and the liver is firm on palpation.
Diagnostic Evaluation
Early diagnosis is critical to the child with EHBA; the outcome in children surgically treated before 2 months of age is much better than in patients with delayed treatment. The diagnosis of biliary atresia is suspected on the basis of the history, physical findings, and laboratory studies. Laboratory tests include a complete blood count, serum bilirubin levels, and liver function studies. Additional laboratory analyses, including α1-antitrypsin level, TORCH titers and other intrauterine infections (see p. 380), hepatitis serology, and urine cytomegalovirus, may be indicated to rule out other conditions that cause cholestasis and jaundice. An abdominal ultrasound is usually performed to identify potential causes of extrahepatic obstruction, such as a choledochal cyst. The patency of the extrahepatic biliary system will be demonstrated by a nuclear scintiscan using technetium 99m iminodiacetic acid (99mTc-IDA, or HIDA; HIDA scan). If there is no evidence of radioactive material excreted into the duodenum, biliary atresia is the most probable diagnosis. A percutaneous liver biopsy is probably the most useful method of diagnosing biliary atresia. The definitive diagnosis is further established during an exploratory laparotomy and an intraoperative cholangiogram that demonstrates complete obstruction at some level of the biliary tree.
Prognosis: Untreated biliary atresia results in progressive cirrhosis and death in most children by 2 years of age. The Kasai portoenterostomy improves the prognosis, but is not always a cure. Biliary drainage can often be achieved if the surgery is performed before the intrahepatic bile ducts are destroyed, usually by 8 weeks of age; otherwise the prognosis is poor. Long-term survival has been reported in children who underwent portoenterostomy before 8 weeks of age. However, even with successful bile drainage, many children ultimately develop liver failure and require liver transplantation. Davenport (2005) reports long-term symptom-free success rates of 10% to 15% in children after portoenterostomy. Reports vary for 10-year survival rates from centers in Japan, Europe, and the United States, with 27% to 68% success rates reported for children following portoenterostomy who survive with native liver (i.e., no transplant required) (Davenport, 2005).
The advances in surgical techniques for liver transplantation and the development of immunosuppressive and antifungal drugs have significantly improved the success of transplantation. Surgical techniques and immunosuppression have contributed to 1- and 5-year survival rates of 87% and 77%, respectively, in children who underwent transplant (Hurwitz and Cox, 2007). The major obstacle remains the shortage of suitable infant donors.
In infants with delayed diagnosis, or in children in whom surgery has failed to provide adequate bile drainage, liver disease progresses. Cirrhosis and splenomegaly occur with hypoalbuminemia, ascites, and coagulopathy. Malabsorption of fat and fat-soluble vitamins and malnutrition result in severe growth failure. Retained bile salts and cholesterol further contribute to pruritus (itching) and xanthomas, often requiring the administration of ursodeoxycholic acid. The severity of pruritus intensifies as the jaundice progresses as the result of disease advancement.
Therapeutic Management
Medical management of biliary atresia is primarily supportive. It includes nutritional support with infant formulas that contain medium-chain triglycerides and essential fatty acids. Supplementation with fat-soluble vitamins (A, D, E, K); a multivitamin; and minerals, including iron, zinc, and selenium, is usually required. Aggressive nutritional support in the form of continuous gastrostomy feedings or total parenteral nutrition may be indicated for moderate to severe growth failure; the enteral solution should be low in sodium. Phenobarbital may be prescribed after hepatic portoenterostomy to stimulate bile flow, and ursodeoxycholic acid may be used to decrease cholestasis and the intense pruritus from jaundice. In cases of advanced liver dysfunction, management is the same as in infants with cirrhosis. (See Chapter 33.)
The primary surgical treatment of biliary atresia is hepatic portoenterostomy (Kasai procedure). This surgical procedure involves dissection of the porta hepatis to expose an area through which bile may drain (Fig. 11-22). A Roux-en-Y jejunal limb is then anastomosed to the porta hepatis (a Y-shaped anastomosis performed to provide bile drainage without reflux). This procedure has several variations. In approximately 90% of infants with biliary atresia who are operated on when younger than 8 weeks of age, bile drainage is achieved. Complications after portoenterostomy include ascending cholangitis, cirrhosis, portal hypertension, and GI bleeding. Prophylactic antibiotics are given after the Kasai procedure to minimize the risk of ascending cholangitis.
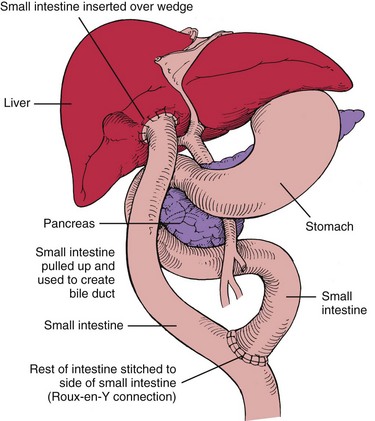
After the Kasai procedure approximately one third of infants become jaundice free and regain normal liver function. Another one third of infants demonstrate liver damage; however, they may be supported by medical and nutritional interventions. A final third require liver transplantation. Liver transplantation is required for children who cannot regain bile flow and for those with end-stage liver disease or severe portal hypertension. Complications after liver transplantation include obstruction and bile leaks at the biliary anastomosis, portal hypertension, hemorrhage, infection, and rejection. Immunosuppressive drugs are required after transplantation.
Nursing Care Management
There are many important nursing interventions for the child with biliary atresia. The nurse should educate family members regarding all aspects of the treatment plan and the rationale for therapy. Immediately after a hepatic portoenterostomy, nursing care is similar to that following any major abdominal surgery. If an interrupted jejunal conduit has been performed, the family needs to learn how to care for the two stomas and how to refeed the bile after feedings. Teaching includes the proper administration of medications. Administration of nutritional therapy, including special formulas, vitamin and mineral supplements, gastrostomy feedings, or parenteral nutrition, is an essential nursing responsibility. Growth failure in such infants is common, and increased metabolic needs combined with ascites, pruritus, and nutritional anorexia constitute a challenge for care. The nurse teaches caregivers how to monitor and administer nutritional therapy in the home. Pruritus may be a significant problem that is addressed by drug therapy or comfort measures such as baths in colloidal oatmeal compounds and trimming of fingernails. The risk of complications of biliary atresia, such as cholangitis, portal hypertension, GI bleeding, and ascites, should be explained to the caregivers.
These children and their families require special psychosocial support. The uncertain prognosis, discomfort, and waiting for transplantation can produce considerable stress. (See Cirrhosis, Chapter 33.) In addition, extended hospitalizations, as well as pharmacologic and nutritional therapy, can impose significant financial burdens on the family, as with any chronic condition. The expertise of a multidisciplinary health care team, including surgeons, gastroenterologists, pediatricians, nurses, nutritionists, pharmacists, child life specialists, and social workers, is often necessary. Parent support groups can be beneficial as well. The Children’s Liver Association for Support Services* and the American Liver Foundation† provide educational materials, programs, and support systems for parents of children with liver disease.
Abdominal Wall Defects
Gastroschisis and omphalocele are two of the more common forms of congenital abdominal wall defects. Gastroschisis occurs in varying incidences worldwide from about 0.4 to 3 in 10,000 births (Ledbetter, 2006), and omphalocele occurs in approximately 1 to 5 in 10,000 live births (Ledbetter, 2006; Stoll, 2007). Numerous reports cite an increase in the incidence of gastroschisis; the cause of this increased incidence is unknown (Islam, 2008). An omphalocele occurs when the abdominal contents herniate through the umbilical ring (hernia of the umbilical cord), usually with an intact peritoneal sac, whereas gastroschisis occurs when the herniation of intestine is lateral to the umbilical ring. This herniation is usually to the right of the umbilicus, and a peritoneal sac is not present.
Omphalocele
Omphalocele is related to a true failure of embryonic development. It occurs when there is failure of the caudal or lateral infolding of the abdominal wall at approximately the third week of gestation. With the deficiency in the abdominal wall, the bowel is unable to complete its return to the abdomen between the tenth and twelfth week of gestation.
The omphalocele is usually covered only by a translucent peritoneal sac (Fig. 11-23). The sac may contain only a small portion of the bowel or most of the bowel and other abdominal viscera, such as the liver. If the sac ruptures, the abdominal contents become exposed. Omphalocele often is associated with other anomalies (50% to 70% incidence of anomalies), including cardiac, neurologic, skeletal, and GU anomalies; imperforate anus; ileal atresia; and bladder exstrophy. Omphalocele is also associated with trisomies 13, 18, and 21 (Down syndrome) and with advanced maternal age (>30 years) (Ledbetter, 2006).
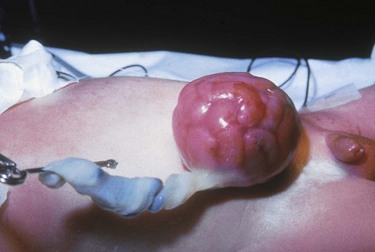
A small omphalocele may go undetected at first glance and appear as a bulge in the umbilical cord. It is therefore imperative to inspect an unusually large umbilical cord for omphalocele before clamping to prevent possible damage to bowel tissue (Donlon, Furdon, and Clark, 2002). (See Care of the Umbilicus, Chapter 8.)
With the increasing frequency of and improvements in prenatal ultrasonography, some abdominal wall defects are being diagnosed prenatally. The benefits of prenatal ultrasonographic diagnosis include the ability to transfer the mother to a tertiary care center, where pediatric surgeons and a neonatal intensive care unit are available to assist with care after delivery.
Initial management after delivery includes inspection of the defect and any associated anomalies. If the bowel covering is intact, a nonadherent dressing is placed over the defect to prevent injury; if the bowel is exposed, the exposed abdominal contents and membranes are covered with a bowel bag or moist dressings and a plastic drape to prevent excessive fluid loss, drying, and temperature instability. IV fluids and antibiotics are administered, and a further evaluation for other associated anomalies is completed. Placement of a Silastic double-lumen catheter (NG-OG) is performed to accomplish gastric bowel decompression.
After initial medical management and stabilization, several surgical options may be carried out depending on the size of the defect, associated medical problems, and surgeon preference. Primary closure of the omphalocele is one option if the defect is small. The sac is resected, contents are reduced into the abdominal cavity, and an attempt is made to close the abdominal fascia with sutures. The abdominal wall may need to be stretched. If an intestinal atresia exists, a bowel resection may be performed, possibly involving a diverting stoma.
When primary closure of the defect is not possible because of the small size of the abdominal cavity or an extremely large omphalocele, staged reduction is accomplished. A silo mesh may be used to house the omphalocele. The silo may be suspended vertically using mild tension. An antibacterial ointment is applied to the silo and suture lines to prevent local infection. Usually the silo is compressed on a daily basis. Once the abdominal cavity is able to accommodate the viscera, the silo is removed and the defect is closed. Every effort is made to accomplish this within 7 to 10 days to minimize the risk of infection. Alternatively some surgeons treat the omphalocele sac with silver sulfadiazine over the ensuing weeks or months until epithelial tissue forms. Repair may occur as late as 1 year if the defect is large (Islam, 2008; Ledbetter, 2006). Another approach involves closure with skin flaps from the lateral abdominal wall.
Postoperatively these infants may require mechanical ventilation and parenteral nutrition. Intraabdominal compression may prevent effective respiration and restrict blood flow to the lower extremities and abdominal organs. Feedings may resume once adequate bowel function is established. Postoperative complications involve many of those discussed below with omphalocele but also include infection, evisceration, intestinal volvulus, obstruction, and a ventral hernia.
Gastroschisis
Gastroschisis occurs when the bowel herniates through a defect in the abdominal wall to the right of the umbilical cord and through the rectus muscle (Fig. 11-24). There is no membrane covering the exposed bowel. Controversy exists regarding the etiology of gastroschisis. It has been suggested that at some point between the bowel’s stay in the umbilical cord and the completion of fixation, a tear occurs at the base of the umbilical cord, allowing the intestine to herniate. The gap between the cord and the tear is filled in by skin, giving the appearance of a defect in the abdominal wall to the right of the umbilical cord. The base of the defect is narrow, and the lack of membranes results in thickening and foreshortening of the bowel. Gastroschisis is usually not associated with other major congenital anomalies (incidence of 10% to 20%); however, jejunoileal atresia, ischemic enteritis, and malrotation may occur as a result of the defect itself. Cryptorchidism in association with gastroschisis has also been reported to range from 24% to 55% (Williams, Butler, and Sundem, 2003). A teratogenic etiology (young maternal age, smoking, alcohol use, acetaminophen, aspirin, and pseudoephedrine) has been suggested as a possible contributor to this defect, as have environmental influences in certain cases (Ledbetter, 2006).
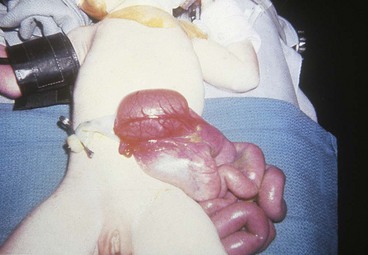
Initial management involves covering the exposed bowel with a transparent plastic bowel bag or loose, moist dressings. If the opening in the abdominal cavity through which bowel is protruding is small and strangulation of bowel is possible, the abdominal opening is enlarged at the bedside. IV fluids and antibiotics are administered, and a double-lumen NG tube is inserted for bowel decompression. Fluid replacement for gastroschisis is increased twofold to threefold because of large losses from the exposed viscera.
Adequate thermoregulation and fluid management are extremely important for both omphalocele and gastroschisis. During surgery the abdominal wall is stretched and the mass of bowel is replaced in the abdomen. If primary closure is not possible, a prefabricated, spring-loaded Silastic silo is placed over the unprotected bowel in labor and delivery or in the neonatal intensive care unit shortly after birth to protect the bowel and decrease fluid loss; primary surgical closure is attempted at a later date. The silo is reduced over several days, at which time it is removed surgically and the defect is closed.
Infants with gastroschisis have traditionally been operated on within 24 hours of birth because of temperature instability, risk of infection in the unprotected bowel, and fluid loss. Studies have shown that outcomes vary in regards to early surgical closure versus silo management and later surgical closure; some outcomes are heavily dependent on the amount of bowel to be replaced into the abdominal cavity and subsequent intraabdominal pressure with primary closure. However, the optimal time for closure has yet to be determined (Islam, 2008).
Postoperatively most infants require mechanical ventilation because of respiratory distress secondary to increased abdominal pressure. Pain management is imperative, especially in the first 72 hours. Morphine and fentanyl are effective opioid analgesics. Many infants also require prolonged nutritional support (parenteral and enteral) because of poor bowel function. Prolonged parenteral nutrition may cause liver failure. Exposure of the bowel to amniotic fluid in utero predisposes the infant to prolonged paralytic ileus and hypomotility. Other complications include infection, transient renal impairment, intestinal obstruction, vena cava compression, and a subsequent decrease in blood flow to the lower extremities.
Prognosis
Advanced surgical techniques, improved parenteral nutrition delivery systems, and better medical management have improved the prognosis for the newborn with an abdominal wall defect. Survival estimates for infants with gastroschisis range from 90% to 95% (Islam, 2008; Ledbetter, 2006). Survival rates for infants with an isolated omphalocele are reported to be 75% to 95% (Heider, Strauss, and Kuller, 2004; McNair, Hawes, and Urquhart, 2006). Because many newborns with omphalocele often have serious associated congenital anomalies, especially cardiac anomalies (40% to 50%), the prognosis for survival of such infants is often not as predictable or as positive as it is for those with gastroschisis (Ledbetter, 2006).
Nursing Care Management
Nursing care is similar to that for any high-risk infant. Infection is a constant threat before surgery, and careful positioning and handling are necessary to prevent rupture of the omphalocele sac or herniated bowel, or disturbance of the Silastic material used for gradual silo reduction. Viscera should be protected with moist dressings or a silo as described previously. Heat and fluid loss from the exposed viscera are major concerns in the preoperative period. Therefore thermoregulation and attention to adequate fluid volume are critical. Fluid replacement is vital and must compensate for losses. The GI tract is decompressed via an NG tube before surgery to aid in bowel reduction.
Postoperative care includes monitoring for signs of complications and assessment of bowel function; pain management with an opioid is also important in the recovery of the infant. Parenteral nutritional support may be necessary when ileus persists. It may require several days or weeks for normal bowel function to return and before full feedings can be achieved. Infants with a prolonged bowel recovery phase are prime candidates for the development of feeding resistance (see Feeding Resistance, Chapter 10); therefore consultation with a feeding specialist in the early postoperative period is recommended to enhance feeding success. Associated long-term problems with gastroschisis include bowel adhesions, bowel obstruction, necrotizing enterocolitis, and moderate to sometimes severe gastroesophageal reflux (Ledbetter, 2006).
Family Support, Discharge Planning, and Home Care: Because these abdominal defects are visible and may be shocking to parents, immediate emotional support at the time of birth is essential. The family needs a brief explanation of the defect and reassurance that their child is in no immediate danger (unless circumstances are different). After the parents have had time to interact with their newborn, inform them about the surgical treatment and postoperative care. At the time of discharge from the hospital, many of these infants are receiving oral feedings, but extended parenteral nutrition may be required if malabsorption and poor bowel function occur. The nurse can ensure continuity of care by referral to a home health care agency, especially if long-term nutritional support is required.
Hernias
A hernia is a protrusion of a portion of an organ or organs through an abnormal opening. The danger of herniation arises when the protrusion is constricted, impairing circulation, or when the protrusion interferes with the function or development of other structures. The herniations discussed in this section are those that protrude through the diaphragm, the abdominal wall, or the inguinal canal.
Congenital Diaphragmatic Hernia
Congenital diaphragmatic hernia (CDH) results when the diaphragm does not form completely, resulting in an opening between the thorax and the abdominal cavity. The most common type of CDH (90%) is a left posterolateral defect, also known as a Bochdalek hernia because the herniation occurs through the foramen of Bochdalek (Fig. 11-25). If the diaphragm does not form completely, the intestines and other abdominal structures, such as the liver, can enter the thoracic cavity, compressing the lung. Lung growth may be arrested on the affected side and to a lesser degree on the contralateral side. Ventilation is further compromised by hypoplasia and compression of the lung, including the airways and blood vessels. In addition to the anatomic defect, pulmonary hypoplasia and pulmonary hypertension have also been recently recognized as components in the pathology of CDH.
Fig. 11-25 A, Normal diaphragm separating the abdominal and thoracic cavities. B, Diaphragmatic hernia with a small lung and abdominal contents in the thoracic cavity. (From Ehrlich PF, Coran AG: Diaphragmatic hernia. In Kliegman RM, Behrman RE, Jenson HB, et al, editors: Nelson textbook of pediatrics, ed 18, Philadelphia, 2007, Saunders.)
This serious defect requires prompt recognition and aggressive treatment to reduce its high mortality. The incidence of CDH is approximately 1 in 2000 to 5000 live births (Ehrlich and Coran, 2007). When stillbirths, intrauterine deaths, and elective abortions are included, the total incidence of CDH is higher than data observed in practice (Brownlee, Howatson, Davis, et al, 2009). Associated anomalies have occurred in as many as 30% of CDH, and CDH is observed in several chromosomal syndromes (Ehrlich and Coran, 2007).
Clinical Manifestations
The most common manifestation of CDH is acute respiratory distress in the newborn. Entrance of air into the intestines after birth further compromises respiration. Infants with a CDH may be dyspneic and cyanotic and have a scaphoid abdomen (because of abdominal contents in the chest). Cardiac output is impaired, and the infant exhibits signs and symptoms of shock. Some infants with small defects may not exhibit respiratory symptoms until later in infancy.
Diagnostic Evaluation
Prenatal diagnosis of CDH as early as the twenty-fifth week of gestation is possible. The three main features detected by ultrasonography that confirm the diagnosis are polyhydramnios, mediastinal shift, and loops of bowel in the chest cavity. In severe cases fetal hydrops is evident. Low MS-AFP levels are seen in cases of CDH; however, the finding is not specific for this anomaly.
Antenatal diagnosis of CDH is reported to have the following advantages: (1) counseling the family regarding pregnancy alternatives and potential problems of the neonatal period; (2) continuation of the pregnancy and further management, including possible antenatal treatment; and (3) transport of the fetus with a CDH in utero to a tertiary center for management. A multidisciplinary team of neonatologists, neonatal nurses, and pediatric surgeons can intervene early in the acute phase to improve the infant’s chances for survival and a positive outcome.
After birth, the diagnosis of CDH may depend on the type of hernia present. In the majority of cases the diagnosis is suspected on the basis of the clinical manifestations and is confirmed by a chest radiograph. The chest radiograph shows fluid- and air-filled loops of intestine in the affected side of the chest. The mediastinum may be shifted to the unaffected side, and auscultation may reveal decreased breath sounds on the affected side.
Therapeutic Management
Fetal Surgery: Advances in fetal diagnostic and surgical techniques led to intrauterine repair of CDH in the 1990s, but the outcomes were poor and prenatal surgery was discontinued for a period in the United States (Kays, 2006). Tracheal occlusion (TO) has been shown to expand the lungs and push the abdominal contents back into the abdomen, thus producing larger, functional lungs. Several surgical techniques, such as PLUG (plug the lung until it grows) and EXIT (ex utero intrapartum treatment), have been used in human fetuses to increase fetal lung growth and establish an effective airway for ventilation. Minimally invasive fetal surgical endoscopic tracheal occlusion techniques (FETENDO) in the United States have had moderate success (Jelin and Lee, 2009). Recently two outcome-prediction factors—intrathoracic liver position and lung (contralateral) area–to–head circumference ratio—have been used in combination with fetal endoscopic TO surgery in Europe with varying results (Deprest, Gratacos, Nicolaides, et al, 2009).
After Birth: Many infants with a CDH require immediate respiratory assistance, which includes endotracheal intubation and GI decompression with a double-lumen catheter to prevent further respiratory compromise. At birth, bag and mask ventilation is contraindicated to prevent air from entering the stomach and especially the intestines, further compromising pulmonary function. In infants with mild respiratory distress, oxygen may be given by hood. However, close attention to the infant’s acid-base status is imperative in the management and prevention of pulmonary hypertension. Low ventilatory positive pressure and the lowest mean airway pressure possible, combined with rapid ventilatory rates (80 to 120 breaths/min), may reduce the incidence of pulmonary leaks from overinflation of the unaffected lung.
IV fluids are initiated during the stabilization period. A transcutaneous oxygen pressure monitor or pulse oximeter may be placed preductally (right hand) and postductally (left hand, arm, or either foot) to monitor the amount of ductal shunting through the patent ductus arteriosus. An umbilical arterial catheter will help monitor postductal arterial oxygen tension (Pao2) and allow infusion of fluids, glucose, and electrolytes. Ductal shunting of deoxygenated blood occurs when pressure in the pulmonary artery is equal to or less than peripheral blood pressure. If pulmonary hypertension is severe with decreased pulmonary venous return, right atrial pressure will be greater than left atrial pressure, resulting in shunting of blood through the foramen ovale. The net results of these events cause further hypoxia, hypercarbia, and acidosis. (See Persistent Pulmonary Hypertension of the Newborn, Chapter 10.)
Because acidosis increases pulmonary hypertension and consequently shunting of unoxygenated blood away from the lungs, it is imperative to monitor acid-base status closely. Close attention to the infant’s thermoregulatory status (maintaining a neutral thermal environment) and glucose requirements during the acute phase is another priority of care. Individualize ventilatory management on the basis of the infant’s response and requirements. Surfactant replacement therapy may also be used to stabilize neonates with CDH, but outcomes have not demonstrated an overall advantage in relation to extracorporeal membrane oxygenation requirement and mortality (Kays, 2006). The use of inhaled nitric oxide to relieve pulmonary hypertension of CDH has also been used in some cases with mixed results (Kays, 2006). (See Nitric Oxide, Surfactant, and Persistent Pulmonary Hypertension of the Newborn, Chapter 10.)
Another strategy that has demonstrated considerable success in the management of CDH is the use of permissive hypercapnia wherein hyperventilation is not employed in order to reduce iatrogenic lung injury and barotrauma. Preductal Spo2 is maintained at 90, Pco2 is ignored, and metabolic acidosis is corrected with buffers instead of hyperventilation. Using lung protective ventilation (gentle ventilation) strategies aimed at decreasing mean inflation pressures (<25cm H2O) and avoiding hyperventilation have demonstrated better overall outcomes and have significantly decreased pulmonary complications such as pneumothorax (Kays, 2006).
Operative treatment involves returning the abdominal organs to the abdomen and repairing the diaphragmatic defect. The timing of surgical repair may vary. Postoperative management involves continuation of ventilatory therapy, monitoring of acid-base balance, and allowing slight hypercapnea. In addition, gastric decompression, thermoregulation, sedation, and maintenance of adequate cardiac output and peripheral perfusion are continued. When muscle paralysis is required, pay careful attention to suctioning oropharyngeal secretions and maintaining intact skin is vital. If paralysis is continued in the postoperative period, appropriate pain management should not be overlooked.
Prognosis: CDH is a complex problem of pulmonary hypoplasia, immature lungs, and other associated problems. The overall mortality rates for CDH are decreasing as the pathophysiology is better understood in relation to current treatment modalities. Current data suggest overall survival rates of 80% to 90% in isolated CDH (Abdullah, Zhang, Sciortino, et al, 2009; Kays, 2006). Surgical repair of the defect alone does not resolve the infant’s problems related to organ immaturity. Long-term complications of CDH include chronic lung disease, gastroesophageal reflux, feeding problems, recurrent diaphragmatic herniation, pneumonia, growth failure, sensorineural hearing loss, scoliosis, and impaired motor and cognitive function.
Nursing Care Management
Assessment of the infant at birth is an integral component of nursing care. This is accentuated in life-threatening cases such as CDH, where prompt recognition of neonatal respiratory distress, cyanosis, a scaphoid abdomen, and a possible mediastinal shift would alert the nurse to investigate further. Any one or a combination of these signs may signal the presence of CDH. A newborn in respiratory distress at birth who does not initially respond to resuscitation is further evaluated for CDH; endotracheal intubation is an option for providing adequate oxygenation until CDH is ruled out. If CDH is diagnosed prenatally and the infant is in distress, endotracheal intubation is required to prevent further accumulation of air in the stomach and intestines and subsequent respiratory compromise.
NURSING ALERT
Any newborn infant with a scaphoid abdomen, moderate to severe respiratory distress, decreased breath sounds unilaterally, and a history of polyhydramnios should be suspected of having a CDH. Ventilation should not be given with bag and mask to prevent further intestinal air and subsequent respiratory compromise.
Preoperative care involves prompt recognition, resuscitation, and stabilization of the infant, including ventilatory support, blood gas monitoring, fluid volume maintenance, and administration of IV fluids and electrolytes. Gastric decompression is achieved with a double-lumen tube, and the infant is observed for signs of impaired cardiac output, acidosis, and hypoxemia.
Postoperative care includes the routine observations discussed in the care of the high-risk infant. Close observation to detect signs of respiratory distress or fluid and electrolyte imbalances is an important nursing function. Closely monitor the infant for signs of mediastinal shift, pulmonary air leak, and infection. Hypovolemia as a result of third spacing of intravascular fluids may occur. Also pay attention to skin care, since these infants often experience prolonged sedation, an increase in skin moisture, tubes and drains coming in contact with the skin, altered nutrition, and altered hemodynamics, all of which place the infant at risk for skin breakdown. Pain management and developmental needs must be met in addition to lifesaving therapy to ensure the infant has optimal development.
Nursing care of the infant with a CDH is also aimed at reducing stimulation either from care activities such as routine suctioning or from environmental factors such as noise and light. Measures that further reduce infant stress, such as management of pain, should be a routine aspect of care for the infant with a CDH.
Because of the serious nature of the condition and the urgency of treatment, the parents are in great need of ongoing support and education regarding postoperative care. The infant with a CDH may require long-term hospitalization and care. As soon as medically possible, the parents should be involved in the daily care of their child.
Umbilical Hernia
The umbilical hernia is a common hernia observed in infants. It occurs when fusion of the umbilical ring is incomplete at the point where the umbilical vessels exit the abdominal wall. It affects African-Americans more often than Caucasians and low-birth-weight and preterm infants more often than full-term infants. An umbilical hernia usually is an isolated defect, but it may be associated with other congenital anomalies, such as Down syndrome (trisomy 21) and trisomies 13 and 18. The size of the defect is variable, and the protrusion is more prominent when the infant is crying (Fig. 11-26). Incarceration, in which the hernia is constricted and cannot be reduced manually, is rare; usually the defect resolves spontaneously by 3 to 5 years of age. If the hernia persists beyond this age, it is usually surgically corrected on an elective basis.
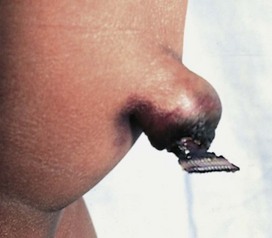
Fig. 11-26 Newborn with umbilical hernia. (From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 5, St Louis, 2007, Mosby.)
Nursing Care Management
The appearance of an umbilical hernia may be disconcerting to parents. Therefore they need reassurance that the defect usually is not harmful. Taping or strapping the abdomen to flatten the protrusion does not aid in resolution and can produce skin irritation.
Nursing care of the child with an umbilical hernia repair is essentially the same as that for other minor GI surgery. The procedure may be performed on an outpatient basis. Observe the child for complications related to a hematoma or infection. The child may resume a normal diet and activity postoperatively; however, strenuous activity or play is restricted for 2 to 3 weeks.
Inguinal Hernia
Inguinal hernias account for approximately 80% of all childhood hernias and occur more frequently in boys than in girls (roughly 6:1). An incidence of 3.5% to 5% is reported in term newborns; 9% to 11% in low-birth-weight and preterm infants, and 30% in very low–birth-weight infants (Aiken and Oldham, 2007).
Pathophysiology
Inguinal hernia comes from persistence of all or part of the processus vaginalis, the tube of peritoneum that precedes the testicle through the inguinal canal into the scrotum (in boys), or the round ligament into the labia (in girls), during the eighth month of gestation. After descent of the testicle, the proximal portion of the processus vaginalis normally atrophies and closes, whereas the distal portion forms the tunica vaginalis, which envelops the testicle in the scrotum. When the upper portion fails to atrophy, the abdominal fluid or an abdominal structure (bowel, ovary, Fallopian tubes) can be forced into it, creating a palpable bulge or mass. The persistent sac may end at any point along the inguinal canal; it may stop at the inguinal ring or extend all the way into the scrotum or labia (Fig. 11-27).
Clinical Manifestations
This common defect is asymptomatic unless the abdominal contents are forced into the patent sac. Most often it appears as a painless inguinal swelling that varies in size. It disappears during periods of rest or is reducible by gentle compression. It appears when the infant cries or strains or when the older child strains, coughs, or stands for a long time. The defect can be palpated as a thickening of the cord in the groin, and the silk glove sign can be elicited by rubbing together the sides of the empty hernial sac.
Sometimes the herniated loop of intestine becomes partially obstructed, producing variable symptoms that may include irritability, tenderness, anorexia, abdominal distention, and difficulty defecating. Occasionally the loop of bowel becomes incarcerated (irreducible), with symptoms of complete intestinal obstruction that, left untreated, will progress to strangulation and necrotic bowel. Incarceration occurs more often in infants under 10 months of age.
Therapeutic Management
The treatment for hernias is prompt, elective surgical repair in the healthy child as soon as the defect is diagnosed. However, an incarcerated hernia requires emergent surgical care. Because there was believed to be a significant incidence of bilateral involvement, many surgeons advocated exploration of both sides; however, this practice has gained disfavor due to complications occurring with open exploration and is seldom used (Brandt, 2008). Laparoscopic exploration of the contralateral side may be performed without risk of injury to the vas deferens (Brandt, 2008).
Nursing Care Management
Prompt recognition of an inguinal hernia is imperative. The hernia may first be noticed when the infant is crying or straining to stool (Valsalva maneuver). Nursing care of the infant or child with an inguinal hernia involves preoperative preparation of the infant and appropriate explanation to the parents of the child’s expected postoperative status. Most hernia repairs can be managed on an outpatient basis. The preterm infant usually has hernia repair several days before discharge. The former preterm infant diagnosed after discharge is admitted the day of surgery and, after repair, is observed for 12 to 24 hours for apnea and bradycardia.
Postoperatively the incision is kept clean and dry, and the infant’s pain is managed appropriately. In infants and small children who are not yet toilet trained, the wound may be covered with an occlusive dressing or left without a dressing. Changing diapers as soon as they become damp helps reduce the chance of irritation or infection of the incision.
No restrictions are placed on the infant’s or toddler’s activity, but older children are cautioned against lifting, pushing, wrestling or fighting, bicycle riding, and athletics for about 3 weeks.
If surgery is postponed, parents need to learn the signs of incarcerated hernia, simple measures to reduce it (a warm bath, avoidance of upright positioning, and comfort measures to reduce crying), and where to call for assistance if relief is not obtained in a reasonably short time.
Femoral Hernia
Femoral hernias are rare in children, with a reported incidence of less than 1% (Brandt, 2008). The incidence is higher in girls than in boys. The hernia may manifest as a recurrent hernia following inguinal hernia repair (Brandt, 2008). Initial symptoms are swelling in the groin area associated with severe abdominal pain and cramping. Treatment and management are the same as for inguinal hernia. Incarceration and strangulation are frequent complications.
Defects of the Genitourinary Tract
External defects of the GU tract are usually obvious at birth. The anatomic location of these defects frequently causes more psychologic concern to parents than does the actual condition or treatment. The timing of medical and surgical procedures for correction of these defects has important implications for children. Surgery involving reproductive organs can be particularly disruptive to preschoolers, who fear punishment, retaliation, body mutilation, or castration. Therefore the trend is toward early correction of visible genital defects, preferably without multiple-stage repairs. Table 11-6 describes renal anomalies, which are typically not obvious at birth.

Phimosis
Phimosis is a narrowing or stenosis of the preputial opening of the foreskin that prevents retraction of the foreskin over the glans penis. It is a normal finding in infants and young boys and usually disappears as the child grows and the distal prepuce dilates. Occasionally the narrowing obstructs the flow of urine, resulting in a dribbling stream or even ballooning of the foreskin with accumulated urine during voiding.
Balanitis is an inflammation or infection of the phimotic foreskin, which occurs occasionally and is managed as any other inflammation or infection. Severe phimosis is treated surgically by circumcision.
Nursing Care Management
Proper hygiene of the phimotic foreskin in infants and young boys consists of external cleansing during routine bathing. The foreskin should not be forcibly retracted, since it may create scarring that can prevent future retraction. Furthermore, retraction of the tight foreskin can result in paraphimosis, a condition in which the retracted foreskin cannot be replaced in its normal position over the glans. This causes edema and venous congestion created by constriction by the tight band of foreskin—a urologic emergency that requires immediate evaluation.
Hydrocele
Hydrocele is the presence of fluid in the processus vaginalis and is a result of the same developmental process as inguinal hernia (see Fig. 11-27, F). A hydrocele in which the upper segment of the processus vaginalis has been obliterated but the tunica vaginalis still contains peritoneal fluid is called a noncommunicating hydrocele. This type of hydrocele is common in newborns and often subsides spontaneously as fluid is gradually absorbed.
A communicating hydrocele is one in which the processus vaginalis remains open and into which peritoneal fluid may be forced by intraabdominal pressure and gravity. The length of the hydrocele depends on the length of the processus vaginalis; it may extend into the tunica vaginalis within the scrotum. The hydrocele is asymptomatic except for a palpable bulge in the inguinal or scrotal area. Unlike a hernia, the hydrocele may not be reducible and may not be produced by a sudden increase in intraabdominal pressure (such as straining). The scrotum appears to be larger after an active day and smaller in the morning. Because a communicating hydrocele represents a patent processus vaginalis, it can predispose the child to herniation. Therefore surgical repair is indicated if spontaneous resolution does not take place by 1 year of age.
Cryptorchidism (Cryptorchism)
Cryptorchidism is failure of one or both testes to descend normally through the inguinal canal into the scrotum. Absence of testes within the scrotum can be a result of (1) undescended (cryptorchid) testes, (2) retractile testes, or (3) anorchism (absence of testes). Undescended testes can be categorized further according to location:
Abdominal—Proximal to the internal inguinal ring
Canalicular—Between the internal and external inguinal rings
Ectopic—Outside the normal pathways of descent between the abdominal cavity and the scrotum
The incidence of cryptorchidism is reported to be as high as 45% in preterm boys and less than 5% in full-term boys; by the age of 1 year the incidence decreases to less than 2% and does not change thereafter (Sijstermans, Hack, Meijer, et al, 2008).
Pathophysiology
Testicular development is influenced by a number of genes, but the dominant one is located on the Y chromosome. This gene stimulates the medullary sex cords of the embryonic gonad to differentiate into secretory Sertoli cells. Beginning around week 7, these cells secrete a glycoprotein, müllerian inhibiting substance, that leads to development of a male genital system. Testicular descent is a critical element of the development of the male genital system. This descent occurs in two phases; the first is dominated by müllerian inhibiting substance and the second phase by testosterone. Between weeks 8 and 15 a cordlike structure, the gubernaculum, extends from the developing testis (located in the lower abdomen) to the labioscrotal swelling. The fetus grows, but the length of this gubernaculum remains relatively fixed, anchoring the testis to the developing inguinal canal (transabdominal migration) in preparation for the second phase of descent. This second phase begins around weeks 25 to 30 and is characterized by shrinkage of the gubernaculum under the influence of testosterone, causing the testis to migrate down the inguinal canal and into a scrotal position (transinguinal migration). Descent is also characterized by protrusion of peritoneum, the processus vaginalis, that closes before birth.
Cryptorchidism occurs when one or both testes fail to descend through the inguinal canal and into the scrotum. Several processes may slow or arrest testicular descent, including endocrinologic abnormalities affecting the hypothalamic-pituitary-testicular axis, denervation of the genitofemoral nerve, traction of the gubernaculum, abnormal development of the epididymis, or preterm birth. Cryptorchid testes are often accompanied by congenital hernias and abnormal testes, and they are at risk for subsequent torsion.
An ectopic testis emerges outside the inguinal ring into the perineum or femoral area, or lies in a transverse scrotal or prepenile location. The most common site is the superficial inguinal pouch. Ectopia is postulated to occur because of obstruction of the scrotal inlet, scarring (fibrosis) of the gubernaculum, or other mechanical anomalies.
Anorchism is the complete absence of a testis. Anorchism is suspected whenever one or both testes cannot be palpated in the patient with apparent cryptorchidism. In some cases, bilateral anorchism is associated with genotypic and phenotypic abnormalities, but it is commonly associated with a normal karyotype (46,XY) and normal genital development. This observation supports the hypothesis that, in most cases, anorchism represents degeneration rather than agenesis of the testes (vanishing testes or testicular regression syndrome).
The cryptorchid or ectopic testis must be differentiated from anorchism because of the risk for malignant degeneration and subfertility when the testis is left in an extrascrotal location. This differentiation may be resolved by an imaging study, such as an ultrasound, or it may require laparoscopic or direct surgical exploration.
Retractile testes can be found at any level within the path of testicular descent, but they are most commonly identified in the groin. Fortunately, they are not truly cryptorchid. Instead, they are introverted to an inguinal or abdominal position because of an overactive cremasteric reflex. The cremasteric reflex, observed as withdrawal of the testis above the scrotum and into the inguinal canal in response to various stimuli, including exposure to cool temperatures, is active during infancy and peaks around age 4 to 5 years. Unlike the cryptorchid testis, the retractile testis can be gently moved into the scrotum without residual tension and does not require treatment.
Clinical Manifestations
A nonpalpable testis is typically observed by the parent or detected during routine physical examination by a nurse practitioner or physician. If one testis is not palpable, the affected hemiscrotum will appear smaller than the other. With bilateral nonpalpable testes, both hemiscrota appear small. In the case of retractile testes, the parents may report intermittently observing the testes in the scrotum, interspersed with periods when they cannot be visualized or palpated. Frequently, the retractile testis will be observed in the scrotum when child is being bathed in warm water.
Diagnostic Evaluation
It is important to differentiate the true undescended testis from the more common retractile testis. Retractile testes can be “milked” or pushed back into the scrotum, but truly undescended ones cannot. For examination, the nurse can obviate the cremasteric reflex by placing the child in a squatting position or by applying firm finger pressure on the external ring before palpating the abdomen or genitalia. (See Fig. 6-39.)
Undescended testes are palpable along the inguinal canal, but those in the abdominal cavity usually are not. Ultrasonography, CT, MRI, and abdominal laparoscopy are sometimes used to verify cryptorchidism in children undergoing orchiopexy. Laparoscopy is the most accurate means for locating nonpalpable testes (Gatti and Ostlie, 2007). Suggestions to employ in diagnostic examination include:
• An undescended testis is usually smaller and softer than its descended mate.
• A well-developed rugous scrotum usually indicates normal testicular descent (may be confused by the presence of a hydrocele or inguinal hernia).
• Retractile testes are usually bilateral (the cremasteric reflex is equally brisk on both sides).
• A testis can usually be distinguished from a lymph node by its elastic nature. A testis is mobile and can be massaged down into the scrotum, although it will spring back into the canal.
• Application of soap, cornstarch, or talcum powder to the tip of the examiner’s fingers facilitates massaging the inguinal canal.
Acquired undescended testes in children who have had normally descended testes are relatively uncommon. Evaluation of the testes should continue to be a part of the routine physical assessment.
Therapeutic Management
Retractile testes that can be manipulated into the scrotum will eventually assume a satisfactory scrotal position without medical or surgical intervention. The diagnosis is not made at a single examination, and parents are asked if they have observed the testes in the scrotum at some time. If so, the anomaly probably represents the retractile variety and the parents can be reassured. By 1 year of age, retractile testes will descend spontaneously in approximately 75% of cases in both full-term and preterm infants. In contrast, true undescended testes rarely descend spontaneously after 1 year of age.
A trial of hormone therapy with luteinizing hormone–releasing hormone (nasal spray) and human chorionic gonadotropin (injection) may be attempted. This method is commonly used in Europe; however, a recent review of the evidence does not support the use of hormonal therapy to elicit testicular descent (Gapany, Frey, Cachat, et al, 2008). Surgical treatment is the preferred management in the United States. If the testes do not descend spontaneously, orchiopexy is performed before the child’s second birthday, preferably between 1 and 2 years of age. Surgical repair is done to (1) prevent damage to the undescended testicle by exposure to the higher degree of body heat in the undescended location, thus maintaining future fertility; (2) decrease the incidence of malignancy formation, which is higher in undescended testicles; (3) avoid trauma and torsion; (4) close the processus vaginalis; and (5) prevent the cosmetic and psychologic handicap of an empty scrotum. Because of the increased propensity toward neoplastic changes (even after orchiopexy), cryptorchid testes are better observed in the scrotal position, where they can be routinely palpated.
The timing of the surgery is important, as it is in any genital surgery. Orchiopexy is usually performed between 6 and 24 months of age. Fewer psychologic effects and a higher rate of fertility may be achieved when repair takes place at an early age. Having both testes in the scrotum by school age prevents psychologic problems related to body image and peer-group embarrassment, since the empty scrotum is smaller in size and altered in shape.
In the routine surgical procedure for undescended testes, the testes are brought down into the scrotum and secured in that position without tension or torsion. A simple orchiopexy for a palpable testis can usually be performed in an outpatient surgical unit. Diagnostic laparoscopic surgery through the umbilicus is expected to improve outcomes in males with cryptorchid testes (Onal and Kogan, 2008). Intraabdominal testes require considerable surgical skill because of technical problems resulting from variations in the length of the spermatic cord, and overnight hospitalization may be necessary. In most cases the family can be reassured of normal testicular function in adulthood.
Nursing Care Management
Postoperative nursing care is directed toward preventing infection and instructing parents in home care of the child, including pain control. Infection is prevented by carefully cleansing the operative site of stool and urine. Observation of the wound for complications and activity restrictions are discussed. The child should avoid vigorous sports activities and use of toys that are straddled for 2 weeks postoperatively to prevent dislodgment of the testis from the scrotum. Parents are concerned about the child’s future fertility, and the nurse counsels the family regarding the prognosis and the optimum time for discussing it with the child—ideally, as a part of sex education. Follow-up care for all boys with cryptorchidism is suggested, especially those in whom orchiopexy was delayed until after age 10 or 11 years or never performed, since these boys are nearly six times more likely than other boys to develop testicular cancer (Walsh, Dall’Era, Croughan, et al, 2007; Gapany, Frey, Cachat, et al, 2008). They should learn how to perform testicular self-examination.
Hypospadias
Hypospadias is a condition in which the urethral opening is located below the glans penis or anywhere along the ventral surface (underside) of the penile shaft (Fig. 11-28). The incidence of hypospadias is reported to be 1 out of 250 to 300 live births, with 10% to 15% of affected newborns having a first-degree male relative (sibling or father) with the same condition (Bukowski and Zeman, 2001; Gray and Moore, 2009). In mild cases the meatus is just below the tip of the penis. In the most severe malformations the meatus is located on the perineum between the halves of the scrotum (bifid scrotum). Chordee, or ventral curvature of the penis, results from the replacement of normal skin with a fibrous band of tissue and usually accompanies more severe forms of hypospadias. In addition, the foreskin is usually absent ventrally and, when combined with chordee, gives the organ a hooded and crooked appearance. In severe cases the altered appearance may leave the infant’s gender in doubt at birth because the perineal position of the meatus may be mistaken for a female urethra. Because undescended testes may also be present, the small penis may appear to be an enlarged clitoris. In any case of ambiguous genitalia, further study, such as chromosome analysis, is essential. (See Disorders of Sex Development, p. 457.)
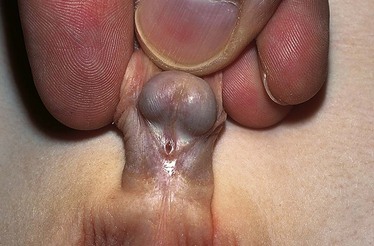
Fig. 11-28 Hypospadias. (Courtesy H. Gil Rushton, MD, Children’s National Medical Center, Washington, DC.)
Surgical Correction
The principal objectives of surgical correction are (1) to enhance the child’s ability to void in the standing position with a straight stream, (2) to improve the physical appearance of the genitalia for psychologic reasons, and (3) to preserve a sexually adequate organ. Many procedures have been described that accomplish one or more of these goals. The choice of surgical procedure is affected primarily by the severity of the defect and the presence of associated anomalies.
Distal hypospadias (a defect noted on the glans penis) may be corrected by a meatal advancement and glanuloplasty (MAGPI) procedure or glans approximation procedure (GAP). The MAGPI requires a dorsal meatotomy with incision of the tissue between the urethral meatus and the glanular groove. The dorsal epithelium is then advanced distally, and care is taken to avoid disrupting the urethra. The ventral skin is then approximated, redundant tissue is carefully excised, and the defect is closed. The GAP requires approximation of the nonjoined urethral opening with lengthening of the urethra and deepithelialization of skin at the lateral edges of the defect. Although the procedure is relatively simple, the potential for urethrocutaneous fistula is significant.
More proximal hypospadias defects typically require alternative approaches. When sufficient ventral skin is present, a skin flap is mobilized that is large enough to reach the tip of the penis. The flap is freed with as much subcutaneous tissue attached as is feasible. Urethral reconstruction is accomplished by reversing the meatal-based flap distally to the glans penis. Specific strategies are used to provide adequate vascular supply to the mobilized skin grafts and to prevent fistula formation. When ventral skin volume is deficient, an island flap repair may be used. This procedure requires more extensive use of skin grafts to create the new urethra and close the defect.
The chordee that often coexists with a hypospadias defect is repaired by release of the ventral skin. When the chordee persists, additional procedures may be indicated. In many cases, a single operation will be all that is needed to correct hypospadias and chordee.
Increased surgical experience and improvements in technique have reduced the number of staged procedures needed for hypospadias defects. However, a staged procedure is indicated in particularly severe defects with marked deficits of available skin for mobilization of flaps and in rare cases when scrotal transposition occurs.
The preferred time for surgical repair is 6 to 12 months of age, before the child has developed body image. Occasionally a short course of testosterone is administered preoperatively to achieve additional penile size to facilitate the surgery.
Nursing Care Management
The nurse should examine every male newborn carefully for hypospadias. If the nurse suspects even mild hypospadias, this is reported to the practitioner. Traditionally the foreskin has been used to repair the hypospadias; however, newer surgical techniques do not require an intact foreskin for repair to be successful. In male infants who remain uncircumcised, the nurse should verify that the newborn’s meatal opening is present on the glans penis, preferably without forcefully retracting the foreskin. Preparation of parents for the type of procedure to be done and the expected cosmetic result helps avert problems. Frequently parents are informed of what is to be surgically corrected but are not advised of what to expect as a reasonable consequence. More refined surgical techniques performed by surgeons specializing in pediatric urologic conditions have improved cosmetic and functional outcomes in these boys. If children are old enough to understand what is occurring, the nurse also prepares them for the operation and the expected outcome.
Hypospadias repair may require some type of urinary diversion with a silicone stent or feeding tube to promote optimum healing and to maintain the position and patency of the newly formed urethra. Sedation may be required for the excessively irritable or restless child, and pain is controlled with analgesics. Epidural anesthesia using a local anesthetic and/or analgesic may be used as an adjunct to general anesthesia for the repair and left in place for postoperative pain management.
The nurse teaches the parents to care for the indwelling catheter or stent and irrigation technique if indicated. They need to know how to empty the urine bag and how to avoid kinking, twisting, or blockage of the catheter or stent. Often the child is discharged with a catheter or stent emptying directly into the diaper. To prevent infection, a tub bath should be avoided until the stent has been removed. An antibacterial ointment may be applied to the penis daily for infection control. In older children a urine collection device can be used. Parents need to learn how to secure the drainage bag to the leg to allow the child to be mobile, and they should be cautioned to never clamp off a catheter. A larger volume bedside urine bag should be used at night to prevent overfilling. Positioning the bag below the waist allows for proper drainage. An extra bag is sent home with the family in case of tears or leakage. The family should encourage the child to increase fluid intake. The child should avoid straddle toys, sandboxes, swimming, and rough activities until allowed by the surgeon.
Epispadias and Exstrophy Complex
Epispadias is a defect of the urinary system characterized by failure of urethral canalization. Bladder exstrophy is a more severe defect characterized by externalization of the bladder, splaying of the urethra with failure of tubular formation, and diastasis (separation) of the pelvic bone (Figs. 11-29 and 11-30). Both these defects are part of a complex of congenital anomalies of GU development that range from relatively mild defects (such as glandular epispadias, or a defect on the dorsal surface [topside] of the penile shaft) to severe lower abdominal defects involving multiple organ systems (such as cloacal exstrophy). Fortunately, the incidence of exstrophy and epispadias complex anomalies is small. Bladder exstrophy, the most common of the defects, is estimated to occur in 1 of 50,000 live births; the defect is just as common in males and females (Nelson, Dunn, and Wei, 2005).
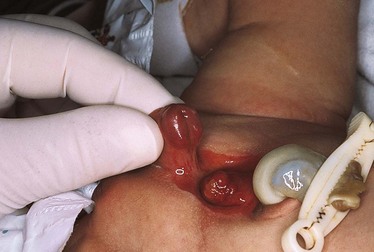
Fig. 11-29 Newborn with bladder exstrophy and epispadias. (Courtesy Tim Yankee, St. Francis Hospital, Tulsa.)
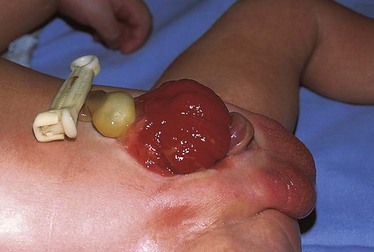
Fig. 11-30 Exstrophy of bladder. (Courtesy H. Gil Rushton, MD, Children’s National Medical Center, Washington, DC.)
Pathophysiology
Exstrophy results from failure of the abdominal wall and underlying structures, including the ventral wall of the bladder, to fuse in utero. As a result, the lower urinary tract is exposed and the everted bladder appears bright red through the abdominal opening. This is accompanied by a constant seepage of urine from the exposed ureteral orifices, making the area malodorous and susceptible to infection. The constant accumulation of urine on the surrounding skin produces tissue ulceration and further infection. Progressive renal damage from infection and obstruction may cause renal failure if left untreated.
In males the defect is almost always associated with epispadias and may include other problems, such as undescended testes and inguinal hernias. Even after reconstruction the sexual handicap in males may be severe because the penis is shortened and does not hang dependently. In females the genitalia may be affected, with a cleft or bifid clitoris; a bifid uterus; completely separated labia; and a duplicate, exstrophic, or absent vagina. In either sex, separation of the pubic bones is generally corrected by pelvic osteotomy, resulting in a normal gait for these children (Nelson, King, Sponseller, et al, 2006). The upper urinary tract is usually normal, and fertility is possible in females but more complex in males, who may require assisted reproductive techniques such as percutaneous sperm aspiration and intracytoplasmatic sperm injection to achieve a pregnancy (D’Hauwers, Feitz, and Kremer, 2008).
Therapeutic Management
The objectives of treatment are (1) preservation of renal function, (2) attainment of urinary control, (3) adequate reconstructive repair for psychologic benefit, (4) prevention of urinary tract infections, and (5) preservation of optimum external genitalia with continence and sexual function. The correction of an epispadias or bladder exstrophy defect is complex and in the past has required multiple surgical procedures and ongoing management of the urinary system. Single-stage surgical closure of bladder exstrophy and epispadias repair with total mobilization is now being performed with varying degrees of success in infants who meet single-stage closure repair criteria (Gearhart, 2001; Husman, 2006; Mitchell, 2005). Widespread acceptance of this procedure began in 1998; therefore the full extent of its success or complications are not yet known (Husman, 2006).
For the child with exstrophy undergoing staged repair, the bladder is closed during the neonatal period, preferably within the first 1 to 2 days of life. Until closure is performed, the bladder is covered with clear plastic wrap or a thin film dressing without adhesive. Petroleum jelly is avoided because it tends to damage the bladder mucosa. After bladder closure, the neonate is monitored for urinary output and for signs of urinary tract or wound infection. At the time of closure, the pelvic diastasis is corrected with an osteotomy or an external fixation device to prevent the waddling gait that occurs when pelvic bone defects are not addressed. Inguinal herniorrhaphies may be completed with initial closure to reduce the risk of subsequent incarceration, particularly for boys.
During the next 3 to 5 years, urine drains freely from the urethra, which has no sphincter mechanism. This period should allow the bladder to gain capacity while the child grows and matures before subsequent surgical repair. The parents should learn to recognize the signs of urinary tract infection and monitor the urinary system regularly via urinalysis and ultrasonographic imaging.
The second stage of exstrophy management is repair of the epispadias and creation of a urethral sphincter mechanism. Epispadias repair attempts to lengthen and straighten the penis to a more dependent position and provide a distal urethra adequate for urination and ejaculation. Creation of a urethral sphincter mechanism requires reconstruction of the bladder neck and tubulization of the proximal urethra to accomplish these goals.
In some children, reconstruction (tightening) of the bladder neck may not provide sufficient resistance to achieve urinary continence. In these cases, suburethral collagen injections or implantation of an artificial urinary sphincter may be performed. Occasionally the bladder fails to achieve an adequate functional capacity, and augmentation enterocystoplasty is required. This procedure is typically combined with the creation of a Mitrofanoff appendiceal stoma because catheterization is particularly difficult after reconstruction of the proximal urethra (see p. 404).
Abnormalities of the genitalia are addressed to ensure optimal sexual function. In boys the testes are typically cryptorchid, and bilateral orchiopexy is combined with reconstruction of the bifid scrotum to preserve testicular function. In girls, surgical enlargement of the vaginal introitus may be needed to permit intercourse. In both genders, plastic surgery to reduce scarring of the genital area or to create an umbilicus may significantly improve the child’s body image and emerging sexual identity.
Nursing Care Management
Physical care of the unrepaired defect includes meticulous hygiene of the bladder area to prevent infection and excoriation of the surrounding tissue. A sterile, nonadherent, moist dressing is placed over the exposed bladder area to prevent infection and to keep the diaper from adhering to the mucosa. Plastic wrap may be placed loosely over the dressing to protect the site and prevent drying of the dressing. A moisture barrier ointment may be prescribed for the surrounding skin to protect it from the constantly draining urine. After bladder closure, if external compression is used to immobilize the pelvis, the skin is inspected periodically for evidence of pressure necrosis.
Fluid management is critical because of the large insensitive water losses from the exposed viscera (Mercy and Brady-Fryer, 2004); therefore IV access is considered early. An umbilical arterial catheter may be inserted for blood specimen withdrawal and infusion of fluids (Mercy and Brady-Fryer, 2004).
Other aspects of preoperative care are similar to those for any major abdominal surgery. If a sterile specimen is needed for evaluation of existing infection, urine may be obtained by aspirating the specimen with a sterile syringe.
Nursing care after the complete primary repair is aimed at decreasing pain and agitation in the infant, preserving pelvic immobility, maintaining ureteric catheter patency, and maintaining the operative site intact. Abdominal distention from crying or other sources is avoided. NG tube decompression may be used to prevent abdominal distention. Dehiscence may occur postoperatively, and signs of suture separation or wound problems are promptly reported to the practitioner. The infant may require mummy wrapping or a spica cast, whereas the older child may have an external fixator in place to achieve pelvic stability and immobilization postoperatively (Nelson, King, Sponseller, et al, 2006). Skin care and care of the external fixation device to ensure appropriate function are essential components of nursing care of the child with exstrophy repair. Chapter 39 discusses nursing care of the child in a spica cast.
Postoperative nursing care after bladder neck reconstruction and antireflux surgery (ureteral reimplantation) includes routine wound care and careful monitoring of urinary output from the bladder and ureteral drainage tubes. Care after a penile lengthening, chordee release, and urethral reconstruction is similar to care after hypospadias repair.
Children who fail to attain urinary continence after bladder neck reconstruction are offered a continent diversion. Preoperatively, a bowel prep is required. In addition to routine postsurgical care, nursing after a continent diversion includes wound care, observation of NG suction (surgery requires bowel resection), and measurement and observation of urinary output. CIC is used to regularly empty the urinary reservoir. Most children are able to learn self-catheterization by age 6 or 7 years. Adult supervision is needed to ensure the child is compliant.
Family Support and Home Care: One of the most devastating aspects of exstrophy of the bladder is its appearance. Although the actual physical care is not difficult, it is not easy for parents to assume responsibility for what to them seems an enormous task because of the emotional impact of the defect.
The nurse should instruct parents regarding a realistic outcome of surgery because unrealistic expectations of the cosmetic and functional result may leave them disappointed and discouraged. Parents often worry about the child’s sexual adjustment, even though they may not voice such concerns. Part of the nursing admission history is directed toward evaluating the parents’ (and child’s, if appropriate) expectation of the surgical repair, knowledge of the possibility of eventual urinary diversion, and feelings concerning this permanent change in body function.
When the infant is discharged with an unrepaired defect, plastic wrap is placed over the defect and diapers are changed frequently to prevent infection, ulceration, and odor. Parents learn to recognize the signs of urinary tract infection (see Chapter 30) and to report a suspected infection to the practitioner. General infant care remains unchanged except for sponge baths rather than immersion in water.
Even with improved reconstructive surgery for these patients, substantial psychologic support and guidance are needed to help them adjust to their fears of inadequate penile size; appearance of the genitalia; potential inability to procreate; and rejection by peers, especially the opposite sex. Ongoing discussion groups for parents and children are particularly useful in resolving these fears and promoting optimum psychologic adjustment, particularly during adolescence.
Obstructive Uropathy
Structural or functional abnormalities of the urinary system can obstruct the normal flow of urine and compromise function. The area proximal to the site of obstruction is exposed to increased intraluminal pressure, dilation, and urinary stasis. For example, when the bladder outlet is obstructed, the renal pelves and both ureters may become distended, a condition called ureterohydronephrosis. However, if one ureterovesical junction is obstructed, the entire ureter and renal pelvis of that side will be affected and the contralateral system will remain normal in appearance and function. Similarly, if the ureteropelvic junction (UPJ) becomes obstructed, the renal pelvis and calyces will become dilated, a condition called hydronephrosis.
In children with end-stage renal disease, the most common types of obstructive uropathy reportedly include posterior urethral valves (PUVs); UPJ; obstructive megaureter, a type of urethral hypoplasia or atresia; ureterocele; and miscellaneous forms of obstruction (Hinds, 2004).
Obstruction may be congenital or acquired, unilateral or bilateral, complete or incomplete, and chronic or acute (Fig. 11-31). Boys are affected more commonly than girls, but obstructive uropathy is suspected whenever a child experiences a congenital urinary system defect. Oligohydramnios (decreased amount of amniotic fluid, usually defined as <500 ml) is often an indication of poor renal function in the fetus. Appropriate fetal pulmonary development is dependent on adequate renal function, and renal impairment may cause fetal pulmonary hypoplasia with resulting respiratory distress at birth (Hinds, 2004; Wu and Johnson, 2009). The leading causes of mortality in obstructive uropathy are pulmonary hypoplasia and preterm birth (Wu and Johnson, 2009). Table 11-7 summarizes common obstructive sites in children and their nursing implications.
TABLE 11-7
COMMON CAUSES OF OBSTRUCTIVE UROPATHY IN CHILDREN
| OBSTRUCTIVE SITE | DESCRIPTION AND NURSING IMPLICATIONS |
| Calyx | Congenital infundibular stenosis, intrinsic blockage from a stone, inflammation, or tumor, causing calyceal dilation or diverticulum; obstructed calyx typically asymptomatic but clinically significant when it serves as a reservoir for infection |
| Ureteropelvic junction | Intrinsic stenosis or extrinsic blockage cause by anomalous blood vessel, kink, or fibrous band; often detected on prenatal ultrasonography or when diuresis causes acute-onset flank pain; radionuclide scan used to determine severity of obstruction and subsequent treatment |
| Ureterovesical junction | Congenital megaureter (congenital obstruction from unknown cause); acquired intrinsic blockage from stone, tumor, or inflammation; or extrinsic obstruction from tumor; megaureter may be asymptomatic or may cause urinary tract infection, hematuria, or abdominal mass |
| Bladder and urethra | Low bladder wall compliance or blockage of bladder outlet caused by bladder neck contracture or hypertrophy, urethral valves, urethral polyp; congenital urethral obstruction detected by observing infant’s initial urination; straining or failure to urinate within first 12-24 hr of life indicative of potentially serious urinary system obstruction |
| Functional obstruction | Vesicosphincter dyssynergia caused by spinal anomalies (such as myelodysplasia or spinal injury) or functional causes (Hinman syndrome) often leading to ureterohydronephrosis, vesicoureteral reflux, and chronic voiding dysfunction |
Pathophysiology
The pathophysiologic changes produced by obstruction are influenced by the location and severity of the blockage and by the presence of complicating factors, such as infection. When the kidney is obstructed, the papilla is flattened; the distal nephron is dilated; and, if the obstruction persists, glomerular filtration is greatly diminished or arrested. Unless the obstruction is relieved, these changes become irreversible, leading to renal insufficiency and atrophy of the affected kidney.
Several factors may magnify the destructive changes associated with obstruction. During the neonatal period the immature kidneys have a higher vascular resistance than do the kidneys of the older child or adult. This condition, combined with the immaturity of the parenchyma, accelerates the irreversible renal damage in response to obstruction. Infection also magnifies the destructive changes associated with urinary obstruction. In contrast to the adult, pyelonephritis in the developing kidney of the infant or child increases the chances of renal scarring that reduces the potential for further growth. However, predicting the extent of scarring and subsequent risk for further damage in every child with pyelonephritis is impossible.
Fortunately, when obstruction is restricted to one kidney, compensatory growth occurs in the contralateral kidney. In the infant and child, compensatory growth includes both hypertrophy (enlargement of existing nephrons) and hyperplasia (replication of new cells). Still, this cannot completely compensate for the loss of renal function created by obstruction of one kidney.
Obstruction also affects the smooth muscle of the renal pelvis, ureter, and bladder. Obstruction of the ureter and renal pelvis causes dilation in an attempt to overcome the obstruction by stronger peristaltic contractions. The process of dilation ultimately leads to urinary stasis and loss of smooth muscle tone unless the obstruction is relieved. The detrusor smooth muscle is also affected by obstruction of the bladder outlet. Like the ureter, the bladder initially responds by increasing the force and duration of its contractions. However, when the obstruction persists, the bladder wall becomes trabeculated and the effectiveness of the detrusor contractions is compromised. In addition to these changes, obstruction of the bladder causes neurologic changes that predispose the bladder to unstable (hyperactive) contractions and subsequent urge incontinence.
Clinical Manifestations
The clinical manifestations of obstructive uropathy depend on the location of the obstructing lesion, its severity, and the underlying cause. Maternal oligohydramnios may signal a renal problem in the developing fetus. Occasionally, UPJ obstruction is diagnosed when a child experiences flank pain, hematuria, and nausea after mild trauma or a urinary tract infection. However, these conditions are more frequently diagnosed on routine prenatal ultrasonographic examination or when a mass is observed during routine newborn examination. Congenital ureteral obstruction also may be asymptomatic, or the obstruction may cause urinary tract infections or an abdominal mass. In contrast, obstruction of the renal pelvis, ureter, or UPJ because of a urinary calculus produces a characteristic pain called renal colic. This pain is characterized by discomfort in the flank, lower back, or lower abdomen. The discomfort is typically intense and it is not relieved by changes in position. Renal colic typically occurs in the early morning hours and persists until the stone passes or is removed. Narcotic analgesia is frequently required to manage the pain produced by an obstructing stone.
Obstruction of the bladder produces lower urinary tract symptoms. These symptoms are closely related to those produced by other dysfunctional voiding conditions, including detrusor instability and urinary retention. Symptoms include poor force of urinary stream, intermittency of voided stream, feelings of incomplete bladder emptying, and postmicturition dribble. In addition, children with obstruction of the bladder or urethra may experience frequency of urination, nocturia, nocturnal enuresis, or urgency to urinate. Urge incontinence may occur, particularly when obstruction causes unstable contractions of the detrusor muscle.
Diagnostic Evaluation
Imaging studies are used to determine the level of the obstruction and the severity of the associated uropathy. Ultrasonography is used to define the anatomy of the obstruction, and radionuclide studies are frequently completed to determine the impact of the obstruction on renal function. Occasionally an IV pyelogram is used to provide a detailed evaluation of urinary tract function, but radionuclide scans or ultrasonography is generally preferred because it exposes the child to less radiation and carries no risk of adverse contrast reactions. The diethylenetriamine pentaacetic acid (DTPA) or mercaptoacetyltriglycine (MAG3) renal scan provides an estimate of renal function as well as renal and ureteral anatomy. The dimercaptosuccinic acid (DMSA) renal scan provides a superior method for determining individual kidney function, but this technique does not allow visualization of the renal pelves, ureters, or bladder. The VCUG is commonly performed to rule out reflux or to evaluate the posterior urethra in boys.
Prenatal detection of obstructive uropathy by sonogram and subsequent fetal surgery to relieve the obstruction in utero, thus preventing further damage as the kidneys develop, has met with varying success; in many cases renal function was not significantly improved (Chevalier, 2004; Salam, 2006). However, others report fewer fetal complications and morbidity with fetal surgical treatment of obstructive uropathy in large centers with more experience performing such procedures (Hofmann, Becker, Meyer-Wittkopf, et al, 2004; Wu and Johnson, 2009).
Therapeutic Management
The management of obstructive uropathy depends on the magnitude of the obstruction and the likelihood that renal function will be compromised unless aggressive intervention is undertaken. For example, UPJ obstruction causing hydronephrosis in the neonate may or may not require surgical intervention. In contrast, PUV obstruction requires aggressive intervention to prevent progressive, severe obstructive uropathy affecting the entire urinary system.
Whenever feasible, obstruction is treated by a surgical procedure that directly ablates the obstructive lesion. For example, UPJ obstruction is ideally treated by pyeloplasty or pyelotomy, urethral valves by endoscopic ablation, or obstructing calculi by extracorporeal or endoscopic shock-wave lithotripsy. Urinary flow is temporarily diverted during the postoperative period via a urethral catheter, ureteral stent, or pyelostomy tube (tube inserted directed into the renal pelvis) until postoperative edema subsides and adequate urinary outflow is achieved.
In contrast, transient or permanent urinary diversion may be required for the child with severe obstructive uropathy causing renal insufficiency who is not able to undergo surgery or who has irreversible damage to the lower urinary tract, posing an ongoing threat to renal function. Transient diversion can be created at the level of the renal pelvis (pyelostomy), the ureters (ureterostomy), or the bladder (vesicostomy), depending on the level of obstruction. The stomas created from these procedures are not typically pouched in the neonate. Instead, the infant’s diaper may be placed higher on the abdomen or a two-diaper system may be used to ensure urinary containment. Permanent urinary diversion usually involves a continent urinary diversion that incorporates a segment of bowel, stomach, or ureter to increase bladder capacity. In contrast to transient diversions, this procedure is reserved for older children and requires ongoing intermittent catheterization.
Prognosis: The prognosis depends on the type of obstruction, the degree of irreversible renal damage, whether renal dysplasia is present, the age at diagnosis, and the severity of complications. Despite improvements in corrective surgery, some patients develop renal failure, which may evolve over a highly variable period that can extend into adulthood. Renal failure can result from hypoplasia-dysplasia, pyelonephritic scarring, and other proposed mechanisms that cause progressive nephron loss. Careful follow-up care should extend throughout childhood and adolescence, especially when any degree of renal insufficiency is present.
Nursing Care Management
Nursing goals in urinary tract obstruction include helping to identify cases, assisting with diagnostic procedures, and caring for children with complications. (See Chapter 30.) Preparing parents and children for procedures, especially urinary diversion procedures, is a major nursing responsibility. (See Preparation for Diagnostic and Therapeutic Procedures, Chapter 27.)
Parents and children need emotional support and counseling during the lengthy management of these disorders. Parents are the primary target during infancy and early childhood, when most reparative surgery is performed. They need assistance in managing the care of the child and in detecting subtle signs of urinary tract infection or renal failure. Parents may perform intermittent catheterizations in the home. Anticholinergics may also be used to decrease spasticity of smooth muscle of the bladder, and parents should be aware of the side effects of these medications (Hinds, 2004). Improved surgical techniques allow for the creation of urinary diversion systems that can be catheterized with greater ease, avoiding mechanical devices, which are often difficult to manage and impede normal everyday activities. (See also Management of Genitourinary Function, p. 403.)
Children with external diversional systems need psychologic support and guidance, especially as they reach adolescence and become more concerned with body image. Those with progressive renal deterioration may face the prospect of dialysis or transplantation and the emotional turmoil that accompanies these procedures.
Disorders of Sex Development (Ambiguous Genitalia)
Until recently, an infant born with ambiguous genitalia was referred to as having an intersex condition. Major advances in the identification of molecular genetic causes of abnormal sexual development, with heightened awareness of ethical issues and patient advocacy concerns, led to reexamination of the nomenclature. In 2006 the term disorders of sex development was proposed to indicate congenital conditions with atypical development of chromosomal, gonadal, or anatomic sex (Lee, Houk, Ahmed, et al, 2006).
The birth of a child with ambiguous genitalia constitutes a crisis quite different from that of other congenital anomalies. Uncertain gender is a potential lifetime social tragedy for the child and family. Furthermore, the electrolyte disturbances that accompany conditions such as congenital adrenal hyperplasia can be life threatening. The identification of appropriate gender must be done with precision and accuracy. The assignment of gender is a complex process; the newborn should be examined by a multidisciplinary team that includes a geneticist, a pediatric urologist, an endocrinologist, a pediatrician, a pediatric psychiatrist, and a pediatric surgeon. Although a rapid assignment of gender helps alleviate some of the parents’ anxiety, an erroneous gender determination requiring a later change is even more stressful for the family. The parents may be advised to tell relatives and friends that the newborn has a congenital malformation of the external genitalia that will require some time for physicians to determine the child’s sex.
Etiology
Genetic sex is determined at the time of conception and depends on whether the ovum is fertilized by a sperm bearing an X chromosome or one bearing a Y chromosome. The phenotypic evidence of gender depends on whether subsequent processes proceed normally: differentiation of primitive gonads, differentiation and development of internal duct systems, and differentiation and development of external genitalia. The normal order of events can be altered by abnormalities of the chromosomal complement, defects of embryogenesis, or biochemical (hormonal) abnormalities. Disturbances in any of these processes will lead to abnormal development evidenced by ambiguous genitalia at birth.
Normal Genitalia and Reproductive Organ Development: For the first 6 weeks of life the developing embryo is morphologically asexual, neither male nor female. The primitive, bipotential (able to form either a testicle or an ovary) gonad consists of an outer layer (the cortex) and an inner medulla. Differentiation into testes or ovaries takes place during the seventh and eighth weeks of gestation. At this time, in the male the medullary portion develops and the cortical zone regresses. In the female the cortex is preserved while the medulla regresses. Active factors from the male testes cause the müllerian duct system to regress. Without these factors the primitive gonad has an inherent tendency to feminize. The embryonic ovary develops in the absence of male hormone stimulation.
The final stage of genital and reproductive organ development is differentiation of the external genitalia, which in the early embryo consists of a urogenital sinus, two lateral labioscrotal swellings, and an anteriorly situated genital tubercle. Depending on the presence or absence of male hormones, the genital tubercle differentiates into a penis or a clitoris. In response to testicular androgens, the labiosacral folds fuse to form a scrotum and the ventral skin of the penis; the urethral folds form the perineal and penile urethra. Without the influence of masculinizing secretions, the urethral folds do not fuse and instead become the labia minora, the labiosacral folds remain unfused to separate into the labia majora, and the urogenital sinus differentiates into a lower vagina and the vaginal and urethral openings (Fig. 11-32).
Abnormal Genitalia and Reproductive Organ Development: Disturbances in the normal order of events in gender determination produce abnormal genitalia and reproductive organ development with the presence of ambiguous or indeterminate external genitalia at birth. Ambiguous genitalia can be variable and may often closely conform to one gender or the other. In some forms the external sexual structures represent those of a normal male or female, whereas the karyotype is the direct opposite. A situation in which the phenotypic gender differs from the chromosomal gender is a disorder of sex development.
A failure or abnormality in any of the four steps of genital and reproductive organ development can lead to abnormal development in subsequent stages. The mechanisms and sites of defective development include:
Abnormal gender determination—Chromosome abnormalities result in disturbance of secondary sexual characteristics and reproductive organ development. (See Chapter 5.)
Abnormal differentiation of gonads—When induction of the bipotential gonad fails, gender differentiation proceeds in the direction of the female phenotype, regardless of karyotype.
Abnormal differentiation of ductal systems—Biologic inactivity of androgenic male organizer substances or insensitivity of ductal tissue to the action of these substances results in a persistent female duct system, which leads to the presence of a uterus and uterine tubes.
Abnormal secretion of or tissue insensitivity to testicular androgen—Complete failure of male hormone secretion produces female external genitalia in a genetic male. Partial or incomplete failure results in incomplete masculinization with ambiguity of the external genitalia. The genetic female fetus exposed to large amounts of androgenic hormone may exhibit varying degrees of masculinization of the external genitalia (congenital adrenal hyperplasia).
Types of Abnormalities
Some disorders with abnormal genital development are not characterized by ambiguous genitalia in the newborn period. For example, the most common sex chromosome disorders do not become apparent until later childhood, adolescence, or even young adulthood, when the individual seeks medical attention because of problems of delayed development or infertility. The four conditions producing ambiguous genitalia in the newborn that require prompt and accurate evaluation are the masculinized female, the incompletely masculinized male, the presence of both male and female sexual organs, and mixed gonadal dysgenesis.
Ambiguous genitalia in the newborn is often a result of virilization in the female by adrenal androgens after the time of early gonadal differentiation. The most common type, congenital adrenogenital hyperplasia (CAH), is an inherited deficiency of adrenal corticoid hormones. (See Chapter 38.) The resulting decrease in cortisol stimulates pituitary secretion of adrenocorticotropic hormone, which causes the adrenal cortex to increase production of adrenal hormones, including the androgens. Because the adrenal gland differentiates later than the gonadal duct system but before differentiation of the external genitalia, masculinization of the external genitalia is the predominant feature. The internal female anatomy is normal. Because even minor illnesses such as vomiting, diarrhea, and dehydration in the child with CAH can lead to life-threatening electrolyte disturbances, CAH should be considered in any situation where the child’s gender is doubtful.
The external genitalia in the incompletely masculinized male may be incompletely male, ambiguous, or completely female. The complex nature of virilization offers numerous opportunities for disturbance in the process. Defects may be a result of deficient production of fetal androgen, deficiency in any of the enzymes needed for testosterone biosynthesis, or unresponsiveness or subresponsiveness of genital structures to testosterone. Individuals who may be either genetic males or females with both ovarian and testicular tissues, with an ovary on one side and a testis on the other or a combination of ovotestis, are rare. The external genitalia may be male (possibly cryptorchid with a micropenis) or normal female, but are ambiguous in the majority of cases.
Mixed gonadal dysgenesis, in which affected infants are sex chromosome mosaics, is the second most common disorder. (See Sex Chromosome Aneuploidies, Chapter 5.) Genitalia vary greatly, but in those who appear predominantly female, the dysplastic testis may cause masculinization at puberty. Table 11-8 describes the external appearance of the genitalia.

Diagnostic Evaluation
Box 11-8 outline diagnostic tools and the significant findings that help determine sex and assist in making a gender assignment.
Therapeutic Management
The management of children with disorders of sex development has evolved in recent years as the traditional approach of assigning gender immediately followed by early surgical reconstruction has been challenged (Karkazis, 2006). Data describing long-term adjustment and quality-of-life issues for individuals with disorders of sex development are lacking. However, anecdotal reports from affected adults have influenced the standard of care. Decision making has become more complex now that endocrine, genetic, social, psychologic, and ethical elements of sex assignment have been integrated into the process. This multidisciplinary team approach, which includes parental participation, has led to the current practice of assigning gender while avoiding irreversible surgical interventions, realizing some children may change gender later in life.
The overall goal of management is to enable the affected child to grow into a well-adjusted, psychosocially stable person who is able to identify with the assigned gender and is content with same (Allen, 2009; Houk, Hughes, Ahmed, et al, 2006; Nabhan and Lee, 2007). Current recommendations for gender assignment involve a number of factors, including:
• The age at presentation is critical. Gender identity is believed to be established by 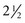 years of age; therefore changing the child’s gender beyond this age is not recommended.
years of age; therefore changing the child’s gender beyond this age is not recommended.
• Male sexual orientation may be in part determined by the amount of androgen exposure in utero; therefore extent of brain virilization should be evaluated.
• Females with CAH, or overvirilization, are often successfully managed with steroids and surgical reconstruction of female genitalia. Prader staging (scale that reflects degree of virilization of the external genitalia) is essential (Allen, 2009).
• Males with severe hypospadias and cryptorchidism (undervirilization) may be successfully reared as males, and surgical repair of both defects has been successful.
• The child with mixed gonadal dysgenesis may be assigned a gender on the basis of phallic size, androgen exposure, and potential for surgical reconstruction; however, long-term studies are lacking as to the success of such management (Lerman, McAleer, and Kaplan, 2000).
• Male infants with a micropenis may respond to testosterone and be successfully reared as boys with the possible exception of those with cloacal exstrophy and penile agenesis (Lerman, McAleer, and Kaplan, 2000). However, a study suggests that even children with cloacal exstrophy may declare themselves as being male during puberty despite female gender assignment (surgically and socially) in the neonatal period (Reiner and Gearhart, 2004).
Clearly the decision for gender assignment in cases of ambiguous genitalia is a difficult one, and the “one size fits all” approach is not applicable. In some cases children may reach puberty and request gender reassignment (Phornphutkul, Fausto-Sterling, and Gruppuso, 2000; Migeon, Wisniewski, Brown, et al, 2002). Federman (2004) emphasizes that evidence in long-term studies suggests that a hormonal role in the sexualization of the brain is more significant than previously recognized. Each child must be considered individually with adequate input from family and extensive diagnostic evaluation. Families often require long-term counseling and follow-up to ensure the child’s welfare and security in the assigned gender.
An evidence-based consensus statement on the management of disorders of sex development has been published and assists in providing a standard of care for these disorders, emphasizing quality of life and patient-centered decision making (Lee, Houk, Ahmed, et al, 2006).
Nursing Care Management
Families need a great deal of support and encouragement from nurses and other members of the health care team to cope with this emotionally charged situation.* Parents are confused, anxious, and overwhelmed by feelings of guilt and shame. They may pressure the health care team for immediate gender assignment because they are concerned about the child and the child’s future and because they must face questioning relatives and friends. The best approach is honesty. The disorder should be treated as any other disorder, with no attempt to camouflage the problem. The sequence of embryologic events leading to the defect can be explained using correct terminology to describe abnormalities of genitalia and reproductive organs. An understanding of the anomaly assists parents in explaining the defect to others, just as with any other physical defect. It requires sympathy and understanding to deal with parental anxiety during this trying period and to guide them throughout the long-term management. (See Chapter 22.)
Key Points
• Congenital malformations or anomalies, or “birth defects,” are present at birth and are a result of genetic or nongenetic influences.
• Typical reactions of parents to an infant with a physical defect include grief over loss of a perfect child, shock, and withdrawal.
• The nurse’s primary roles in care of an infant with a physical defect are caregiver, provider of family support, and supplier of information.
• Surgery initiates a number of physiologic responses, including cardiovascular, respiratory, endocrine, renal, GI, immune, neurologic, and fluid and electrolyte.
• One of the largest groups of congenital anomalies includes those associated with the embryonic neural tube, the most common of which are SB and myelomeningocele.
• When taken before and during pregnancy, folic acid supplementation may prevent as many as 50% to 70% of the cases of NTDs, anencephaly, and SB.
• Care of the infant and child with myelomeningocele requires both immediate and long-term professional supervision. Associated problems include infection, neurologic damage, impaired renal function, musculoskeletal impairment, and latex allergy.
• Hydrocephalus is a symptom of an underlying brain pathologic condition, demonstrated by impaired absorption of CSF or obstruction to the flow of CSF within the ventricles.
• Therapy for hydrocephalus involves relief of the ventricular dilation, treatment of the underlying brain pathologic condition, prevention and/or treatment of complications, and management of problems related to psychomotor development.
• Treatment of DDH involves maintaining the head of the femur correctly positioned in the acetabulum by means of an external device, usually the Pavlik harness.
• Treatment of clubfoot involves manual overcorrection of the deformity, maintenance of the correction until normal muscle balance is gained, and follow-up observation to detect possible recurrence of the deformity.
• CL deformities are repaired at the earliest opportunity; CP repair may be delayed to take advantage of growth changes.
• Management of CP involves a multidisciplinary approach involving professionals from surgery, medicine, nursing, social work, dentistry, speech-language pathology, and audiology.
• Major nursing challenges with infants born with either CL or CP involve feeding.
• TEF is an abnormal connection between the esophagus and the trachea, placing the untreated infant at risk for life-threatening pulmonary aspiration.
• Anorectal defects are often associated with other congenital anomalies, such as those involving the GI tract and kidneys.
• CDH may be diagnosed in the first trimester of pregnancy and usually causes moderate to severe respiratory distress at birth.
• Umbilical and inguinal hernias are common in children and require minor surgical intervention with excellent postoperative recovery.
• Abdominal wall defects, omphalocele and gastroschisis, require careful nursing care involving thermoregulation, fluid management, and prevention of infection preoperatively and postoperatively.
• GU defects are repaired early to promote normal function and psychosocial adjustment.
• In disorders of sex development, gender assignment is established after careful evaluation of prenatal and postnatal influences, karyotype, genitalia features, surgical possibilities, future fertility, and potential sexual function.
References
Abdullah, F, Zhang, Y, Sciortino, C, et al. Congenital diaphragmatic hernia: outcome review of 2,173 surgical repairs in US infants. Pediatr Surg Int. Aug 30, 2009. [(E-pub ahead of print)].
Achildi, A, Grewal, H. Congenital anomalies of the esophagus. Otolaryngol Clin North Am. 2007;40(1):219–244.
Aiken, JJ, Oldham, KT. Inguinal hernias. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
A-Kader, HH, Balistreri, WF. Cholestasis. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Allen, L. Disorders of sexual development. Obstet Gynecol Clin North Am. 2009;36(2):25–45.
American Academy of Pediatrics, Committee on Genetics. Folic acid for the prevention of neural tube defects. Pediatrics. 2007;119(5):1031.
American Academy of Pediatrics, Task Force on Sudden Infant Death Syndrome. The changing concept of sudden infant death syndrome: diagnostic coding shifts, controversies regarding the sleeping environment, and new variables to consider in reducing risk. Pediatrics. 2005;116(5):1245–1255.
Arosarena, OA. Cleft lip and palate. Otolaryngol Clin North Am. 2007;40(1):27–60.
Barker, E, Saulino, M, Caristo, AM. Spina bifida. RN. 2002;66(12):33–38.
Bijnen, CL, Don Griot, PJW, Mulder, W, et al. Tongue-lip adhesion in the treatment of Pierre Robin sequence. J Craniofacial Surg. 2009;20(2):315–320.
Brandt, ML. Pediatric hernias. Surg Clin North Am. 2008;88(1):27–43.
Brown, JP. Orthopaedic care of children with spina bifida: you’ve come a long way, baby!. Orthop Nurs. 2001;20(4):51–58.
Brownlee, EM, Howatson, AG, Davis, CRF, et al. The hidden mortality of congenital diaphragmatic hernia: a 20-year review. J Pediatr Surg. 2009;44(2):317–320.
Buchenau, W, Urschitz, MS, Sautermeister, J, et al. A randomized clinical trial of a new orthodontic appliance to improve upper airway obstruction in infants with Pierre Robin sequence. J Pediatr. 2007;151(2):145–149.
Bukowski, TP, Zeman, PA. Hypospadias: of concern but correctable. Contemp Pediatr. 2001;18(2):89–109.
Buran, CF, McDaniel, AM, Brei, TJ. Needs assessment in a spina bifida program: a comparison of the perceptions by adolescents with spina bifida and their parents. Clin Nurs Spect. 2002;16(5):256–262.
Centers for Disease Control and Prevention. Racial/ethnic differences in the birth prevalence of spina bifida—United States, 1995-2005. MMWR. 2009;57(53):1409–1413.
Centers for Disease Control and Prevention. Folate status in women of childbearing age, by race/ethnicity—United States, 1999-2000, 2001-2002, and 2003-2004. MMWR. 2007;55(51):1377–1380.
Chapman, KL, Hardin-Jones, MA, Goldstein, JA, et al. Timing of palatal surgery and speech outcome. Cleft Palate Craniofac J. 2008;45(3):297–308.
Chevalier, RL. Perinatal obstructive nephropathy. Semin Perinatol. 2004;28(2):124–131.
Chiafery, M. Care and management of the child with shunted hydrocephalus. Pediatr Nurs. 2006;32(3):222–225.
Cleft Palate Foundation. Factsheet: what about breastfeeding? May 2008, The Foundation; available at www.cleftline.org. [(accessed June 2, 2009)].
Dauria, D, Marsh, JL. Mandibular distraction osteogenesis for Pierre Robin sequence: what percentage of neonates need it? J Craniofac Surg. 2008;19(9):1237–1243.
Davenport, M. Biliary atresia. Semin Pediatr Surg. 2005;14(1):42–48.
Dehlink, E, Prandstetter, C, Eiwegger, T, et al. Increased prevalence of latex-sensitization among children with chronic renal failure. Allergy. 2004;59(7):734–738.
de Jong, TP, Chrzan, R, Klijn, AJ, et al. Treatment of the neurogenic bladder in spina bifida. Pediatr Nephrol. 2008;23(6):889–896.
Deprest, JA, Gratacos, E, Nicolaides, K, et al. Changing perspectives on the perinatal management of isolated congenital diaphragmatic hernia in Europe. Clin Perinatol. 2009;36(2):329–347.
DeRoo, LA, Wilcox, AJ, Drevon, CA, et al. First-trimester maternal alcohol consumption and the risk of infant oral clefts in Norway: a population-based case-control study. Am J Epidemiol. 2008;168(6):638–646.
D’Hauwers, K, Feitz, W, Kremer, J. Bladder exstrophy and male fertility: pregnancies after ICSI with ejaculated or epididymal sperm. Fertil Steril. 2008;89(2):387–389.
Donlon, CR, Furdon, SA, Clark, DA. Look before you clamp: delivery room examination of the umbilical cord. Adv Neonat Care. 2002;2(1):19–26.
Doolin, E. Bowel management for patients with myelodysplasia. Surg Clin North Am. 2006;86(2):505–514.
Drake, JM. The surgical management of pediatric hydrocephalus. Neurosurgery. 2008;62(Suppl 2):633–640.
Ehrlich, PJ, Coran, AG. Diaphragmatic hernia. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Faggin, R, Bernardo, A, Stieg, P, et al. Hydrocephalus in infants less than six months of age: effectiveness of endoscopic third ventriculostomy. Eur J Pediatr Surg. 2009;19(4):216–219.
Faulks, S, Luther, B. Changing paradigm for the treatment of clubfeet. Orthop Nurs. 2005;24(1):25–30.
Federman, DD. Three facets of sexual differentiation. N Engl J Med. 2004;350(4):323–324.
Finnell, RH, Gould, A, Spiegelstein, O. Pathobiology and genetics of neural tube defects. Epilepsia. 2003;44(Suppl 3):14–23.
Frey, L, Hauser, WA. Epidemiology of neural tube defects. Epilepsia. 2003;44(Suppl 3):4–13.
Fritsch, MJ, Kienke, S, Ankerman, T, et al. Endoscopic third ventriculostomy in infants. J Neurosurg. 2005;103(Pediatr 1):50–53.
Gapany, C, Frey, P, Cachat, F, et al. Management of cryptorchidism in children: guidelines. Swiss Med Weekly. 2008;138(33-34):492–498.
Gatti, J, Ostlie, D. The use of laparoscopy in the management of nonpalpable undescended testes. Curr Opin Pediatr. 2007;19(3):349–353.
Gearhart, JP. Complete repair of bladder exstrophy in the newborn: complications and management. J Urol. 2001;165(6 Pt 2):2431–2433.
Golden, JA, Bönnemann, CG. Developmental structural disorders. In Goetz CG, ed.: Textbook of clinical neurology, ed. 3, Philadelphia: Saunders, 2007.
Graham, JM, Gomez, M, Halberg, A, et al. Management of deformational plagiocephaly: repositioning versus orthotic therapy. J Pediatr. 2005;146(2):258–262.
Gray, M, Moore, KN. Urologic disorders: adult and pediatric care. St Louis: Mosby; 2009.
Gupta, N, Park, J, Solomon, C, et al. Long-term outcomes in patients with treated childhood hydrocephalus. J Neurosurg. 2007;106(Suppl 5):334–339.
Hassan, FA. Compliance of parents with regard to Pavlik harness treatment in developmental dysplasia of the hip. J Pediatr Orthop B. 2009;18(3):111–115.
Heider, AL, Strauss, RA, Kuller, JA. Omphalocele: clinical outcomes in cases with normal karyotypes. Am J Obstet Gynecol. 2004;190(1):135–141.
Hinds, AC. Obstructive uropathy: considerations for the nephrology nurse. Nephrol Nurs J. 2004;31(2):166–180.
Hofmann, R, Becker, T, Meyer-Wittkopf, M, et al. Fetoscopic placement of transurethral stent for intrauterine obstructive uropathy. J Urol. 2004;171(1):384–386.
Holcomb, GW, 3rd., Rothenberg, SS, Bax, KM, et al. Thoracoscopic repair of esophageal atresia and tracheoesophageal fistula: a multi-institutional analysis. Ann Surg. 2005;242(3):422–428.
Honein, MA. Impact of folic acid fortification of the US food supply and occurrence of neural tube defects. JAMA. 2001;285(23):2981–2986.
Hosalkar, HS, Horn, D, Friedman, JE, et al. The hip. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Hosalkar, HS, Spiegel, DA, Davidson, RS. The foot and toes. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Houk, C, Hughes, I, Ahmed, S, et al. Summary of consensus statement on intersex disorders and their management. Pediatrics. 2006;118(2):753–757.
Hummel, P, Fortado, D. Impacting infant head shapes. Adv Neonat Care. 2005;5(6):329–340.
Hurwitz, M, Cox, KL. Liver transplantation. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Husman, D. Surgery insight: advantages and pitfalls of surgical techniques for the correction of bladder exstrophy. Nat Clin Pract Urol. 2006;3(2):95–100.
Islam, S. Clinical care outcomes in abdominal wall defects. Curr Opin Pediatr. 2008;20(3):305–310.
Jelin, E, Lee, H. Tracheal occlusion for fetal congenital diaphragmatic hernia: the US experience. Clin Perinatol. 2009;36(2):349–361.
Kadrian, D, van Gelder, J, Florida, D, et al. Long-term reliability of endoscopic third ventriculostomy. Neurosurg. 2008;62(Suppl 2):614–621.
Karkazis, K. Early genital surgery to remain controversial. Pediatrics. 2006;118(2):814–815.
Kaufman, BA. Neural tube defects. Pediatr Clin North Am. 2004;51(2):389–419.
Kays, DW. Congenital diaphragmatic hernia and neonatal lung lesions. Surg Clin North Am. 2006;86(2):329–352.
Kelly, DA, Davenport, M. Current management of biliary atresia. Arch Dis Child. 2007;92(12):1132–1135.
Kestle, JR. Pediatric hydrocephalus: current management. Neurol Clin North Am. 2003;21(4):883–895.
Kimata, H. Latex allergy in infants younger than 1 year. Clin Exp Allergy. 2004;34(12):1910–1915.
Kinsman, SL, Johnston, MV. Congenital anomalies of the central nervous system. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Kirkham, C, Harris, S, Grzybowski, S. Evidence-based prenatal care, part I, General prenatal care and counseling issues. Am Fam Physician. 2005;71(7):1307–1316.
Krauss, MJ, Morrissey, AE, Winn, HN, et al. Microcephaly: an epidemiologic analysis. Am J Obstet Gynecol. 2003;188(6):1484–1490.
Lazzaretti, CC, Pearson, C. Myelodysplasia. In Allen PJ, Vessey JA, Schapiro NA, eds.: Primary care of the child with a chronic condition, ed 5, St Louis: Mosby, 2010.
Ledbetter, DJ. Gastroschisis and omphalocele. Surg Clin North Am. 2006;86(2):249–260.
Lee, P, Houk, C, Ahmed, S, et al. Consensus statement on management of intersex disorders. Pediatrics. 2006;118(2):e488–e500.
Lerman, SE, McAleer, IM, Kaplan, GW. Sex assignment in cases of ambiguous genitalia and its outcome. Urology. 2000;55(1):8–11.
Levitt, MA, Peña, A. Anorectal malformations. Orphanet J Rare Dis. 2007;2:33.
Levitt, MA, Peña, A. Outcomes from the correction of anorectal malformations. Curr Opin Pediatr. 2005;17(3):394–401.
Little, J, Cardy, A, Munger, RG. Tobacco smoking and oral clefts: a meta-analysis. Bull World Health Org. 2004;82(3):213–218.
Littlefield, TR, Saba, NM, Kelly, KM. On the current incidence of deformational plagiocephaly: an estimation based on prospective registration at a single center. Semin Pediatr Neurol. 2004;11(4):301–304.
Maher, AB, Salmond, SW, Pellino, TA. Orthopaedic nursing, ed 3. Philadelphia: Saunders; 2002.
Masarei, AG, Wade, A, Mars, M, et al. A randomized control trial investigating the effect of presurgical orthopedics on feeding in infants with cleft lip and/or palate. Cleft Palate Craniofac J. 2007;44(2):182–193.
Matthews, TJ, Trends in spina bifida and anencephalus in the United States, 1991-2006. Hyatsville, Md: National Center on Health Statistics; 2009. available at www.cdc.gov/nchs/products/pubs/pubd/hestats/spine_anen.pdf [(accessed June 1, 2009)].
McNair, A, Hawes, J, Urquhart, H. Caring for the newborn with an omphalocele. Neonat Netw. 2006;25(5):319–327.
Mercy, N, Brady-Fryer, B. Bladder exstrophy: a challenge for nursing care. J WOCN. 2004;31(5):293–298.
Merritt, L. Recognizing craniosynostosis. Neonatal Network. 2009;28(6):369–376.
Merritt, L. Understanding the embryology and genetics of cleft lip and palate, part I. Adv Neonat Care. 2005;5(2):64–71.
Migeon, CJ, Wisniewski, AB, Brown, TR, et al. Ambiguous genitalia with perineoscrotal hypospadias in 46,XY individuals: long-term medical, surgical, and psychosexual outcome. Pediatrics. 2002;110(3):e31.
Mitchell, ME. Bladder exstrophy repair: complete primary repair of exstrophy. Urology. 2005;65(1):5–8.
Mitrofanoff, P, Liard, A. Bladder reconstruction and substitution. In: Gearhart JP, Rink RC, Mouriquand PDE, eds. Pediatric urology. Philadelphia: Saunders, 2001.
Mittler, MA. Time to failure of ventriculoperitoneal shunts (oral abstract). J Neurosurg. 2005;103(Suppl Pediatr 1):A103.
Moore, C, Kogan, BA, Parekh, A. Impact of urinary incontinence on self-concept in children with spina bifida. J Urol. 2004;171(4):1659–1662.
Nabhan, Z, Lee, P. Disorders of sex development. Curr Opin Obstet Gynecol. 2007;19(5):440–445.
Nelson, C, Dunn, R, Wei, J. Contemporary epidemiology of bladder exstrophy in the United States. J Urol. 2005;173(5):1728–1731.
Nelson, C, King, J, Sponseller, P, et al. Repeat pelvic osteotomy in patients with failed closure of bladder exstrophy: applications and outcomes. J Pediatr Surg. 2006;41(6):1109–1112.
Ngo, Q, Ranger, A, Singh, RN, et al. External ventricular drains in pediatric patients. Pediatr Crit Care Med. 2009;10(3):346–351.
Onal, B, Kogan, B. Additional benefit of laparoscopy for nonpalpable testes: finding a contralateral patent processus. Urology. 2008;71(6):1059–1063.
Orenstein, S, Peters, J, Khan, S, et al. Congenital anomalies: esophageal atresia and tracheoesophageal fistula. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Phornphutkul, C, Fausto-Sterling, A, Gruppuso, PA. Gender self-reassignment in an XY adolescent female born with ambiguous genitalia. Pediatrics. 2000;106(1):135–137.
Pope, W. External ventriculostomy: a practical application for the acute care nurse. J Neurosci Nurs. 1998;30(3):185–190.
Reiner, WG, Gearhart, JP. Discordant sexual identity in some genetic males with cloacal exstrophy assigned to female sex at birth. N Engl J Med. 2004;350(4):333–341.
Robinson, S, Proctor, M. Diagnosis and management of deformational plagiocephaly: a review. J Neurosurg Pediatrics. 2009;3(4):284–295.
Rowe, DE, Jadhav, AL. Care of the adolescent with spina bifida. Pediatr Clin North Am. 2008;55(6):1359–1374.
Rudy, C. Hydrocephalus. J Pediatr Health Care. 2005;19(2):111):127–128.
Salam, MA. Posterior urethral valve: outcome of antenatal intervention. Int J Urol. 2006;13(10):1317–1322.
Samaniego, IA. A sore spot in pediatrics: risk factors for pressure ulcers. Pediatr Nurs. 2003;29(4):278–282.
Sijstermans, K, Hack, W, Meijer, R, et al. The frequency of undescended testis from birth to adulthood: a review. Int J Androl. 2008;31(1):1–11.
Stoll, B. The umbilicus. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Stothard, KJ, Tennant, PW, Bell, R, et al. Maternal overweight and obesity and the risk of congenital anomalies: a systematic review and meta-analysis. JAMA. 2009;301(6):636–650.
Sutton, LN. Fetal surgery for neural tube defects. Best Pract Res Clin Obstet Gynaecol. 2008;22(1):175–188.
Tinanoff, N. Cleft lip and palate. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Volpe, JJ. Neurology of the newborn, ed 4. Philadelphia: Saunders; 2001.
Wagner, W, Koch, D. Mechanisms of failure after endoscopic third ventriculostomy in young infants. J Neurosurg. 2005;103(Pediatr 1):43–49.
Walsh, T, Dall’Era, M, Croughan, M, et al. Prepubertal orchiopexy for cryptorchidism may be associated with lower risk of testicular cancer. J Urol. 2007;178(4 Pt 1):1440–1446.
Williams, T, Butler, R, Sundem, T. Management of the infant with gastroschisis: a comprehensive review of the literature. NAINR. 2003;3(2):55–63.
Wolff, T, Witkop, CT, Miller, T, et al. Folic acid supplementation for the prevention of neural tube defects: an update of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2009;150(9):632–639. [W112-W115].
Wu, S, Johnson, MP. Fetal lower urinary tract obstruction. Clin Perinatol. 2009;36(2):377–390.
*223 S. Wacker Drive, Suite 2400, Chicago, IL 60606-4802; 800-221-6827; www.easterseals.com. In Canada, www.easterseals.org.
†1275 Mamaroneck Ave., White Plains, NY 10605; 914-997-4488, 914-997-4488; www.marchofdimes.com. In Canada, www.marchofdimes.ca.
‡800 Celebration Ave., Suite 225, Celebration, FL 34747; 407-566-8304; e-mail: staff@birthdefects.org; www.birthdefects.org.
§1600 Clifton Road, Atlanta, GA 30333; 800-CDC-INFO; www.cdc.gov/ncbddd/index.html.
*Information is available from Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities, Division of Birth Defects and Developmental Disabilities, 1600 Clifton Road NE, MS E-86, Atlanta, GA 30333; 800-CDC-INFO; e-mail: cdcinfo@cdc.gov; www.cdc.gov/ncbddd/folicacid. And from March of Dimes Resource Center, 1275 Mamaroneck Ave., White Plains, NY 10605; www.marchofdimes.com.
*4590 McArthur Blvd., NW, Suite 250, Washington, DC 20007-4226; 202-944-3285, 800-621-3141; fax: 202-944-3295; www.sbaa.org.
*Additional information regarding latex allergy may be found at www.latex-allergy.org, and http://latexallergylinks.tripod.com. For a list of latex products and alternative products, see Latex List on the Spina Bifida Association home page, www.spinabifidaassociation.org.
†Latex-free product lists are available from the American Latex Allergy Association’s online resource manual, available at www.latexallergyresources.org/ResourceManual/section1/index.cfm. American Latex Allergy Association, PO Box 198, Slinger, WI 53086; 888-972-5378; www.latexallergyresources.org.
*12413 Centralia Road, Lakewood, CA 90715-1653; 562-924-6666; www.nhfonline.org.
†870 Market St., Suite 705, San Francisco, CA 94102; 415-732-7040; www.hydroassoc.org. A booklet titled About Hydrocephalus: A Book for Parents is available in English or Spanish; also available is Prenatal Hydrocephalus: A Book for Parents. In Canada, contact the Spina Bifida and Hydrocephalus Association of Canada; 800-565-9488; www.sbhac.ca.
*For additional information contact the Automotive Safety Program, Riley Hospital for Children, Indiana University School of Medicine, 575 West Drive, Room 004, Indianapolis, IN 46202; 317-274-2977 (in Indianapolis), 800-543-6227; www.preventinjury.org.
*1504 E. Franklin St., Suite 102, Chapel Hill, NC 27514-2820; 919-933-9044; e-mail: info@cleftline.org; www.cleftline.org.
†800 Celebration Ave., Suite 225, Celebration, FL 34747; 407-566-8304, www.birthdefects.org.
‡1275 Mamaroneck Ave., White Plains, NY 10605; 914-997-4488, www.marchofdimes.com. In Canada, www.marchofdimes.ca.
*25379 Wayne Mills Place, Suite 143, Valencia, CA 91355; 877-679-8256; www.classkids.org.
†75 Maiden Lane, Suite 603, New York, NY 10038; 212-668-1000; www.liverfoundation.org/education/info/biliaryatresia.
*Some parent and clinical resources may be found at the Accord Alliance website, www.accordalliance.org. DSD Guidelines and Handbook for Parents resources are available at www.dsdguidelines.org.