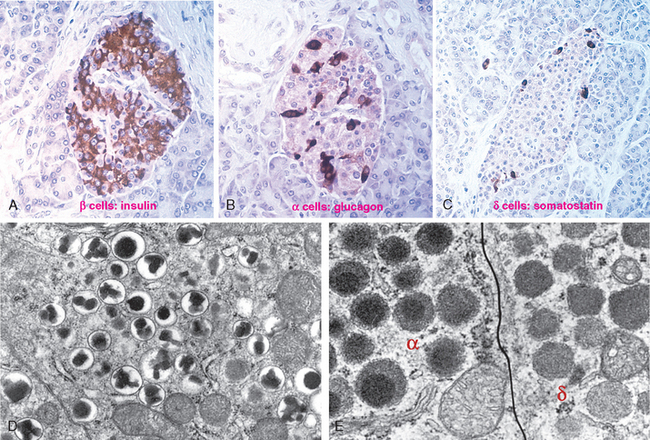
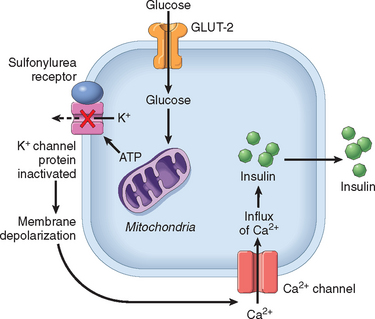
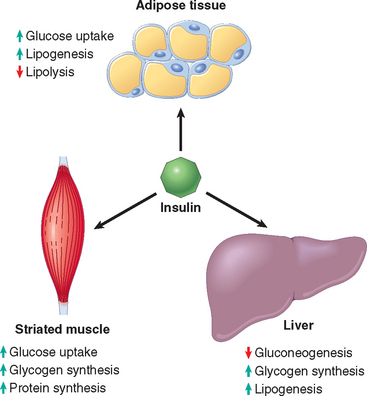
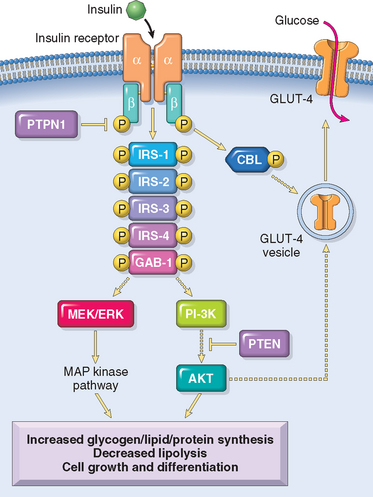
1 Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004;101:613.
2 Asa SL, Ezzat S. The pathogenesis of pituitary tumors. Annu Rev Pathol Mech Dis. 2009;4:97.
3 Karhu A, Aaltonen LA. Susceptibility to pituitary neoplasia related to MEN-1, CDKN1B and AIP mutations: an update. Hum Mol Genet. 2007;16(Spec No 1):R73.
4 Pellegata NS, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15558.
5 Vierimaa O, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006;312:1228.
6 Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064.
7 Devdhar M, et al. Hypothyroidism. Endocrinol Metab Clin North Am. 2007;36:595.
8 Kere J. Overview of the SLC26 family and associated diseases. Novartis Found Symp. 2006;273:2.
9 Gull WW. On a cretinoid state supervening in adult life in women. Transactions of the Clinical Society of London. 1873;7:180.
10 Hashimoto H. Zur Kenntnis der lymphomatösen Veränderung der Schilddrüse (Struma lymphomatosa). Arch Klin. 97, 1912.
11 Kavvoura FK, Akamizu T, Awata T, et al. Cytotoxic T-lymphocyte associated antigen 4 gene polymorphisms and autoimmune thyroid disease: a meta-analysis. The Journal of Clinical Endocrinology and Metabolism. 2007;92:3162.
12 Jacobson EM, Tomer Y. The CD40, CTLA-4, thyroglobulin, TSH receptor, and PTPN22 gene quintet and its contribution to thyroid autoimmunity: back to the future. J Autoimmun. 2007;28:85.
13 McLachlan SM, Nagayama Y, Pichurin PN, et al. The link between Graves’ disease and Hashimoto’s thyroiditis: a role for regulatory T cells. Endocrinology. 2007;148:5724.
14 Graves RJ. A newly observed affection of the thyroid gland in females. London Med Surg J. 1835;7:516.
15 Jacobson EM, Huber A, Tomer Y. The HLA gene complex in thyroid autoimmunity: from epidemiology to etiology. J Autoimmun. 2008;30:58.
16 Baloch ZW, LaVolsi VA. Our approach to follicular-patterned lesions of the thyroid. J Clin Pathol. 2007;60:244.
17 Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer. 2006;6:292.
18 Marques AR, Espadinha C, Catarino AL, et al. Expression of PAX8-PPAR gamma 1 rearrangements in both follicular thyroid carcinomas and adenomas. J Clin Endocrinol Metab. 2002;87:3947.
19 Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev. 2007;28:742.
20 Santarpia L, El-Naggar AK, Cote GJ, Myers JN, Sherman SI. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J Clin Endocrinol Metab. 2008;93:278.
21 Williams ED. Chernobyl and thyroid cancer. J Surg Oncol. 2006;94:670.
22 Albores-Saavedra J, Wu J. The many faces and mimics of papillary thyroid carcinoma. Endocr Pathol. 2006;17:1.
23 Adeniran AJ, Zhu Z, Gandhi M, et al. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am J Surg Pathol. 2006;30:216.
24 Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855.
25 Santoro M, Carlomagno F. Drug insight: Small-molecule inhibitors of protein kinases in the treatment of thyroid cancer. Nat Clin Pract Endocrinol Metab. 2006;2:42.
26 Roodman GD, Dougall WC. RANK ligand as a therapeutic target for bone metastases and multiple myeloma. Cancer Treat Rev. 2008;34:92.
27 Brown EM. Clinical lessons from the calcium-sensing receptor. Nat Clin Pract Endocrinol Metab. 2007;3:122.
28 Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443.
29 Pinhas-Hamiel O, Zeitler P. Acute and chronic complications of type 2 diabetes mellitus in children and adolescents. Lancet. 2007;369:1823.
30 Herman MA, Kahn BB. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J Clin Invest. 2006;116:1767.
31 Youngren JF. Regulation of insulin receptor signaling. Cell Mol Life Sci. 2007;64:873.
32 Imagawa A, Hanafusa T, Miyagawa J, Matsuzawa Y. A novel subtype of type 1 diabetes mellitus characterized by a rapid onset and an absence of diabetes-related antibodies. Osaka IDDM Study Group. N Engl J Med. 2000;342:301.
33 Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661.
34 Todd JA, Walker NM, Cooper JD, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857.
35 Jones EY, Fugger L, Strominger JL, Siebold C. MHC class II proteins and disease: a structural perspective. Nat Rev Immunol. 2006;6:271.
36 Park Y. Functional evaluation of the type 1 diabetes (T1D) susceptibility genes. Diabetes Res Clin Pract. 2007;77(Suppl 1):S110.
37 Chistiakov, et al. The crucial role of IL-2/IL-2RA-mediated immune regulation in the pathogenesis of type 1 diabetes, an evidence coming from genetic and animal model studies. Immunol Lett. 2008;118:1.
38 Hviid A, Stellfeld M, Wohlfahrt J, Melbye M. Childhood vaccination and type 1 diabetes. N Engl J Med. 2004;350:1398.
39 Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med. 1986;314:1360.
40 Knip M, Siljander H. Autoimmune mechanisms in type 1 diabetes. Autoimm Rev. 2008;7:550.
41 Zhang L, et al. Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol. 2008;20:111.
42 Taplin CE, Barker JM. Autoantibodies in type 1 diabetes. Autoimmunity. 2008;41:11.
43 Frayling TM. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. 2007;8:657.
44 Zeggini E, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008.
45 Michael MD, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Molecular Cell. 2000;6:87.
46 Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:193.
47 Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840.
48 Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. 2006;116:1776.
49 Lyssenko V, Lupi R, Marchetti P, et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest. 2007;117:2155.
50 Babenko AP, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355:456.
51 Gloyn AL, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838.
52 Murphy R, et al. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet Med. 2008.
53 Stoy J, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Nat Acad Sci USA. 2007;104:15040.
54 Agostini M, et al. Non-DNA binding, dominant-negative, human PPARgamma mutations cause lipodystrophic insulin resistance. Cell Metab. 2006;4:303.
55 Harja E, et al. Vascular and inflammatory stresses mediate ather-osclerosis via RAGE and its ligands in apoE-/- mice. J Clin Invest. 2008;118:183.
56 Yamamoto Y, et al. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J Clin Invest. 2001;108:261.
57 Clarke M, Dodson PM. PKC inhibition and diabetic microvascular complications. Best Pract Res Clin Endocrinol Metab. 2007;21:573.
58 Tomlinson DR, Gardiner NJ. Glucose neurotoxicity. Nat Rev Neurosci. 2008;9:36.
59 Daneman D. Type 1 diabetes. Lancet. 2006;367:847.
60 Stumvoll M, et al. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365:1333.
61 Boyle PJ. Diabetes mellitus and macrovascular disease: mechanisms and mediators. Am J Med. 2007;120:S12.
62 Zimmet P, et al. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782.
63 Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881.
64 Pi-Sunyer X, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one-year results of the look AHEAD trial. Diabetes Care. 2007;30:1374.
65 Delonlay P, et al. Neonatal hyperinsulinism: clinicopathologic correlation. Hum Pathol. 2007;38:387.
66 Newell-Price J, et al. Cushing’s syndrome. Lancet. 2006;367:1605.
67 Cushing HW. The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism). Bull Johns Hopkins Hosp. 1932;50:137.
68 Mazzuco TL, et al. Aberrant GPCR expression is a sufficient genetic event to trigger adrenocortical tumorigenesis. Mol Cell Endocrinol. 2007;23:265-266.
69 Horvath A, et al. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet. 2006;38:794.
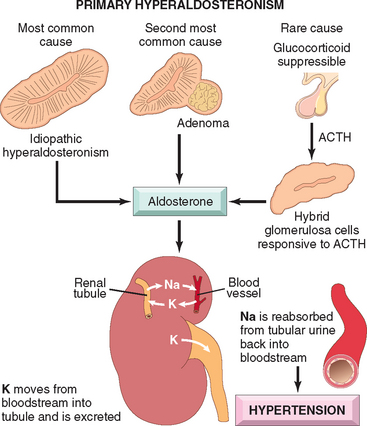
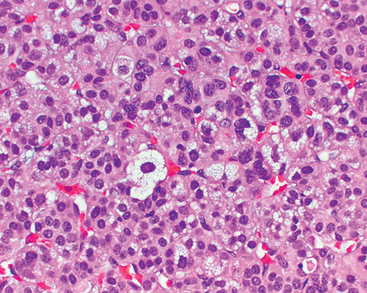
70 Conn JW, Louis LH. Primary aldosteronism: a new clinical entity. Trans Assoc Am Physicians. 1955;68:215.
71 Leopold JA, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nature Medicine. 2007;13:189.
72 Goncalves J, et al. Congenital adrenal hyperplasia: focus on the molecular basis of 21-hydroxylase deficiency. Expert Rev Mol Med. 2007;9:1.
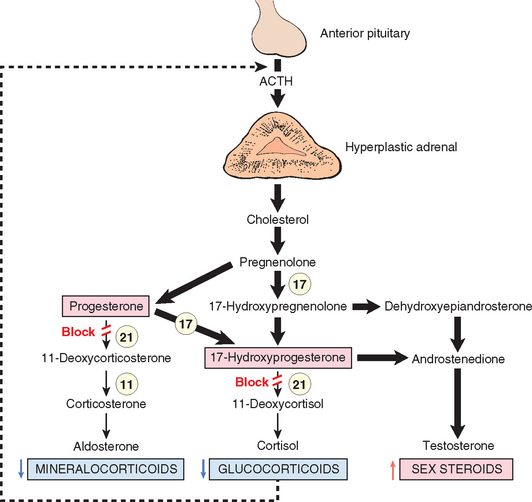
73 Merke DP, et al. Adrenomedullary dysplasia and hypofunction in patients with classic 21-hydroxylase deficiency. N Engl J Med. 2000;343:1362.
74 Waterhouse R. A case of suprarenal apoplexy. Lancet. 1911;1:577.
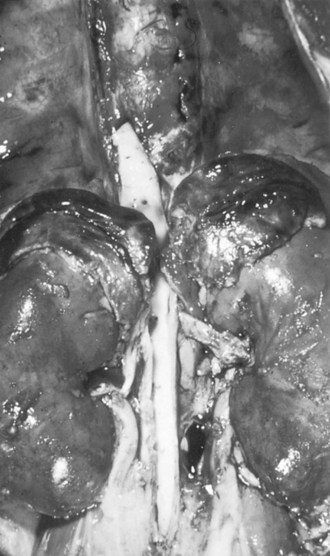
75 Friderichsen C. Nebennierenapoplexie bei kleinen Kindern. Jahrbuch für Kinderhilkunde. 1918;87:109.
76 Addison T. On the constitutional and local effects of disease of the supra-renal capsules. London: Samuel Highley, 1855.
77 Mathis D, Benoist C. A decade of AIRE. Nat Rev Immunol. 2007;7:645.
78 Young WFJr. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007;356:601.
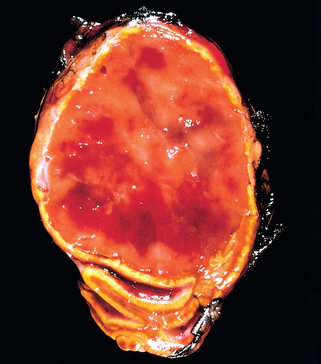
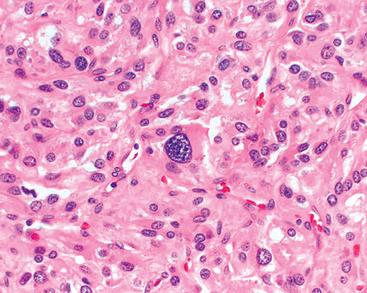
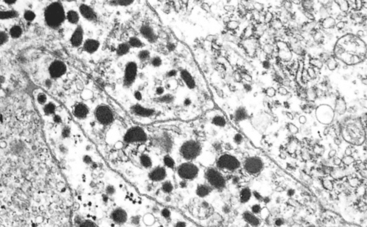
79 Elder EE, et al. Pheochromocytoma and functional paraganglioma syndrome: no longer the 10% tumor. J Surg Oncol. 2005;89:193.
80 Kaelin WG. Von hippel-lindau disease. Annu Rev Pathol. 2007;2:145.
81 Scacheri PC, et al. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet. 2006;2:e51.
82 Agarwal SK, et al. Transcription factor JunD, deprived of menin, switches from growth suppressor to growth promoter. Proc Nat Acad Sci USA. 2003;100:10770.
83 Gujral TS, et al. Molecular mechanisms of RET receptor-mediated oncogenesis in multiple endocrine neoplasia 2B. Cancer Res. 2006;66:10741.