34 The reproductive system
Overview
In this chapter, we describe the endocrine control of the female and male reproductive systems as the basis for understanding drug actions in sex hormone replacement, contraception, treatment of infertility, management of labour and treatment of erectile dysfunction.
Introduction
Drugs that affect reproduction (both by preventing conception and more recently for treating infertility) transformed society in the latter half of the last century. In this chapter, we briefly summarise salient points in reproductive endocrinology as a basis for understanding the numerous important drugs that work on the male and female reproductive systems. Such drugs are used for contraception, to treat infertility, as sex hormone replacement and in obstetric practice to influence labour. The principle of negative feedback is stressed and is central to understanding how hormones interact to control reproduction1—many drugs, including agents used to prevent or assist conception, work by influencing negative feedback mechanisms. The chapter concludes with a short section on erectile dysfunction.
Endocrine Control of Reproduction
Hormonal control of the reproductive systems in men and women involves sex steroids from the gonads, hypothalamic peptides and glycoprotein gonadotrophins from the anterior pituitary.
Neurohormonal Control of the Female Reproductive System
Increased secretion of hypothalamic and anterior pituitary hormones occurs in girls at puberty and stimulates secretion of oestrogen from the ovaries. This causes maturation of the reproductive organs and development of secondary sexual characteristics, and also accelerated growth followed by closure of the epiphyses of the long bones. Sex steroids, oestrogens and progesterone are thereafter involved in the menstrual cycle, and in pregnancy. A simplified outline is given in Figures 34.1 and 34.2.

Fig. 34.1 Hormonal control of the female reproductive system.
The Graafian follicle (GF) is shown developing on the left, then involuting to form the corpus luteum (CL) on the right, after the ovum (•) has been released. FSH, follicle-stimulating hormone; GnRH, gonadotrophin-releasing hormone; LH, luteinising hormone.

Fig. 34.2 Plasma concentrations of ovarian hormones and gonadotrophins in women during normal menstrual cycles.
Values are the mean ± standard deviation of 40 women. The shaded areas indicate the entire range of observations. Day 1 is the onset of menstruation. E and F show diagrammatically the changes in the ovarian follicle and the endometrium during the cycle. Ovulation on day 14 of the menstrual cycle occurs with the mid-cycle peak of luteinising hormone (LH), represented by the vertical dashed line. A, arterioles; FSH, follicle-stimulating hormone; V, venules.
(After van de Wiele R L, Dyrenfurth I 1974 Pharmacol Rev 25: 189–217.)
The menstrual cycle begins with menstruation, which lasts for 3–6 days, during which the superficial layer of uterine endometrium is shed. The endometrium regenerates during the follicular phase of the cycle after menstrual flow has stopped. A releasing factor, gonadotrophin-releasing hormone (GnRH), is secreted from peptidergic neurons in the hypothalamus which discharge in a pulsatile fashion, approximately one burst per hour. GnRH stimulates the anterior pituitary to release gonadotrophic hormones (Fig. 34.1)—follicle-stimulating hormone (FSH) and luteinising hormone (LH). These act on the ovaries to promote development of small groups of follicles, each of which contains an ovum. One follicle develops faster than the others and forms the Graafian follicle (Figs 34.1 and 34.2E), which secretes oestrogens, and the rest degenerate. The ripening Graafian follicle consists of thecal and granulosa cells surrounding a fluid-filled centre, within which lies an ovum. Oestrogens are responsible for the proliferative phase of endometrial regeneration, which occurs from day 5 or 6 until mid-cycle (Fig. 34.2B,F). During this phase, the endometrium increases in thickness and vascularity, and at the peak of oestrogen secretion there is a prolific cervical secretion of mucus of pH 8–9, rich in protein and carbohydrate, which facilitates entry of spermatozoa. Oestrogen has a negative feedback effect on the anterior pituitary, decreasing gonadotrophin release during chronic administration of oestrogen as oral contraception (see below). In contrast, the high endogenous oestrogen secretion just before mid-cycle sensitises LH-releasing cells of the pituitary to the action of the GnRH and causes the mid-cycle surge of LH secretion (Fig. 34.2C). This, in turn, causes rapid swelling and rupture of the Graafian follicle, resulting in ovulation. If fertilisation occurs, the fertilised ovum passes down the fallopian tubes to the uterus, starting to divide as it goes.
Stimulated by LH, cells of the ruptured follicle proliferate and develop into the corpus luteum, which secretes progesterone. Progesterone acts, in turn, on oestrogen-primed endometrium, stimulating the secretory phase of the cycle, which renders the endometrium suitable for the implantation of a fertilised ovum. During this phase, cervical mucus becomes more viscous, less alkaline, less copious and in general less welcoming for sperm. Progesterone exerts negative feedback on the hypothalamus and pituitary, decreasing the release of LH. It also has a thermogenic effect, causing a rise in body temperature of about 0.5°C at ovulation, which is maintained until the end of the cycle.
If implantation of a fertilised ovum does not occur, progesterone secretion stops, triggering menstruation. If implantation does occur, the corpus luteum continues to secrete progesterone, which, by its effect on the hypothalamus and anterior pituitary, prevents further ovulation. The chorion (an antecedent of the placenta) secretes human chorionic gonadotrophin (HCG), which maintains the lining of the uterus during pregnancy. For reasons that are not physiologically obvious, HCG has an additional pharmacological action in stimulating ovulation. As pregnancy proceeds, the placenta develops further hormonal functions and secretes a variety of hormones, including gonadotrophins, progesterone and oestrogens. Progesterone secreted during pregnancy controls the development of the secretory alveoli in the mammary gland, while oestrogen stimulates the lactiferous ducts. After parturition, oestrogens, along with prolactin (see Ch. 32), are responsible for stimulating and maintaining lactation, whereas high doses of exogenous oestrogen suppress this.
Oestrogens, progestogens (progesterone-like drugs), androgens and the gonadotrophins are described below—see Figure 34.3 for biosynthetic pathways.

Fig. 34.3 The biosynthetic pathway for the androgens and oestrogens, with sites of drug action.
(See also Fig. 32.5.) Finasteride is used in benign prostatic hyperplasia, and anastrazole to treat breast cancer in postmenopausal women.
Hormonal control of the female reproductive system 
Neurohormonal Control of the Male Reproductive System
As in women, hypothalamic, anterior pituitary and gonadal hormones control the male reproductive system. A simplified outline is given in Figure 34.4. GnRH controls the secretion of gonadotrophins by the anterior pituitary. This secretion is not cyclical as in menstruating women, although it is pulsatile in both sexes (see below). FSH is responsible for the integrity of the seminiferous tubules, and after puberty is important in gametogenesis through an action on Sertoli cells, which nourish and support developing spermatozoa. LH, which in the male is also called interstitial cell-stimulating hormone (ICSH), stimulates the interstitial cells (Leydig cells) to secrete androgens—in particular testosterone. LH/ICSH secretion begins at puberty, and the consequent secretion of testosterone causes maturation of the reproductive organs and development of secondary sexual characteristics. Thereafter, the primary function of testosterone is the maintenance of spermatogenesis and hence fertility—an action mediated by Sertoli cells. Testosterone is also important in the maturation of spermatozoa as they pass through the epididymis and vas deferens. A further action is a feedback effect on the anterior pituitary, modulating its sensitivity to GnRH and thus influencing secretion of LH/ICSH. Testosterone has marked anabolic effects, causing development of the musculature and increased bone growth which results in the pubertal growth spurt, followed by closure of the epiphyses of the long bones.

Fig. 34.4 Hormonal control of the male reproductive system.
FSH, follicle-stimulating hormone; GnRH, gonadotrophin-releasing hormone; ICSH, interstitial cell-stimulating hormone.
Secretion of testosterone is mainly controlled by LH/ICSH, but FSH also plays a part, possibly by releasing a factor similar to GnRH from the Sertoli cells which are its primary target. The interstitial cells that synthesise testosterone also have receptors for prolactin, which may influence testosterone production by increasing the number of receptors for LH/ICSH.
Behavioural Effects of Sex Hormones
As well as controlling the menstrual cycle, sex steroids affect sexual behaviour. Two types of control are recognised: organisational and activational. The former refers to the fact that sexual differentiation of the brain can be permanently altered by the presence or absence of sex steroids at key stages in development.
In rats, administration of androgens to females within a few days of birth results in long-term virilisation of behaviour. Conversely, neonatal castration of male rats causes them to develop behaviourally as females. Brain development in the absence of sex steroids follows female lines, but is switched to the male pattern by exposure of the hypothalamus to androgen at a key stage of development. Similar but less complete behavioural virilisation of female offspring has been demonstrated following androgen administration in non-human primates, and probably also occurs in humans if pregnant women are exposed to excessive androgen.
The activational effect of sex steroids refers to their ability to modify sexual behaviour after brain development is complete. In general, oestrogens and androgens increase sexual activity in the appropriate sex. Oxytocin, which is important during parturition (see below), also has roles in mating and parenting behaviours, its action in the central nervous system being regulated by oestrogen (see Ch. 32).
Drugs Affecting Reproductive Function
Oestrogens
Oestrogens are synthesised by the ovary and placenta, and in small amounts by the testis and adrenal cortex. The starting substance for synthesis of oestrogen (and other steroids) is cholesterol. The immediate precursors to the oestrogens are androgenic substances—androstenedione or testosterone (Fig. 34.3). There are three main endogenous oestrogens in humans: oestradiol, oestrone and oestriol (Fig. 34.3). Oestradiol is the most potent and is the principal oestrogen secreted by the ovary. At the beginning of the menstrual cycle, the plasma concentration is 0.2 nmol/l, rising to ~2.2 nmol/l in mid-cycle.
Actions
Oestrogen acts in concert with progesterone, and induces synthesis of progesterone receptors in uterus, vagina, anterior pituitary and hypothalamus. Conversely, progesterone decreases oestrogen receptor expression in the reproductive tract. Prolactin (see Ch. 32) also influences oestrogen action by increasing the numbers of oestrogen receptors in the mammary gland, but has no effect on oestrogen receptor expression in the uterus.
Effects of exogenous oestrogen depend on the state of sexual maturity when the oestrogen is administered:
Oestrogens have several metabolic actions, including mineralocorticoid (retention of salt and water) and mild anabolic actions. They increase plasma concentrations of high-density lipoproteins, a potentially beneficial effect (Ch. 23) that may contribute to the relatively low risk of atheromatous disease in premenopausal women compared with men of the same age. However, oestrogens also increase the coagulability of blood, and increase the risk of thromboembolism. This effect is dose related.
Mechanism of action
As with other steroids, oestrogen binds to type 4 (i.e. nuclear) receptors (Ch. 3). There are at least two types of oestrogen receptor, termed ERα and ERβ. Binding is followed by interaction of the resultant complexes with nuclear sites and subsequent genomic effects. In addition to these ‘classic’ intracellular receptors, some oestrogen effects, in particular its rapid vascular actions, may be initiated by interaction with membrane receptors (e.g. Chen et al., 1999). Acute vasodilatation caused by 17-β-oestradiol is mediated by nitric oxide, and a plant-derived (phyto-) oestrogen called genistein (which is selective for ERβ, as well as having quite distinct effects from inhibition of protein kinase C) is as potent as 17-β-oestradiol in this regard. Oestrogen receptor modulators (receptor-selective oestrogen agonists or antagonists) are mentioned briefly below.
Preparations
Many preparations (oral, transdermal, intramuscular, implantable and topical) of oestrogens are available for a wide range of indications. These preparations include natural (e.g. estradiol, estriol) and synthetic (e.g. mestranol, ethinylestradiol, diethylstilbestrol) oestrogens. Oestrogens are presented either as single agents or combined with progestogen.
Pharmacokinetic aspects
Natural as well as synthetic oestrogens are well absorbed in the gastrointestinal tract, but after absorption the natural oestrogens are rapidly metabolised in the liver, whereas synthetic oestrogens are degraded less rapidly. There is a variable amount of enterohepatic cycling, which forms the basis for drug interaction, because broad-spectrum antibiotic use alters bowel flora and can thereby render oral contraception ineffective (Ch. 56). Most oestrogens are readily absorbed from skin and mucous membranes. They may be given as intravaginal creams or pessaries for local effect. In the plasma, natural oestrogens are bound to albumin and to a sex steroid-binding globulin. Natural oestrogens are excreted in the urine as glucuronides and sulfates.
Unwanted effects
Unwanted effects of oestrogens include tenderness in the breasts, nausea, vomiting, anorexia, retention of salt and water with resultant oedema, and increased risk of thromboembolism. More details of the unwanted effects of oral contraceptives are given below.
Used intermittently for postmenopausal replacement therapy, oestrogens cause menstruation-like bleeding. Oestrogen causes endometrial hyperplasia unless given cyclically with a progestogen. When administered to males, oestrogens result in feminisation.
Oestrogen administration to pregnant women can cause genital abnormalities in their offspring. Carcinoma of the vagina was more common in young women whose mothers were given diethylstilbestrol in early pregnancy in a misguided attempt to prevent miscarriage (see Ch. 57).
The clinical uses of oestrogens and antioestrogens are summarised in the box. In addition, see the section below on postmenopausal hormone replacement therapy (HRT).
Oestrogen Receptor Modulator
Raloxifene, a ‘selective oestrogen receptor modulator’ (SERM), has antioestrogenic effects on breast and uterus but oestrogenic effects on bone, lipid metabolism and blood coagulation. It is used for prevention and treatment of postmenopausal osteoporosis (Ch. 35) and also reduces the incidence of oestrogen receptor-positive breast cancer to an extent similar to tamoxifen while causing fewer adverse events (Barret-Connor et al., 2006; Vogel et al., 2006). The US Food and Drug Administration has supported its use to reduce the risk of invasive breast cancer in postmenopausal women with osteoporosis and in postmenopausal women at high risk for invasive breast cancer. Unlike oestrogen, it does not prevent menopausal flushes.
Tamoxifen has antioestrogenic action on mammary tissue but oestrogenic actions on plasma lipids, endometrium and bone. It produces mild oestrogen-like adverse effects consistent with partial agonist activity. The tamoxifen–oestrogen receptor complex does not readily dissociate, so there is interference with the recycling of receptors.
Tamoxifen upregulates transforming growth factor-β, a cytokine that retards the progression of malignancy, and which also has a role in controlling the balance between bone-producing osteoblasts and bone-resorbing osteoclasts (Ch. 35).
Tamoxifen is discussed further in Chapter 55.
Antioestrogens
Antioestrogens compete with natural oestrogens for receptors in target organs; in addition to SERMs (raloxifene, tamoxifen) which are partial agonists in some tissues and antagonists in others, there are drugs that are pure oestrogen receptor antagonists.
Clomiphene inhibits oestrogen binding in the anterior pituitary, so preventing the normal modulation by negative feedback and causing increased secretion of GnRH and gonadotrophins. This results in a marked stimulation and enlargement of the ovaries and increased oestrogen secretion. The main effect of its antioestrogen action in the pituitary is to induce ovulation. It is used in treating infertility caused by lack of ovulation. Twins are common, but multiple pregnancy is unusual.
See the box on oestrogens and antioestrogens for a summary of clinical uses.
Oestrogens and antioestrogens
Clinical uses of oestrogens and antioestrogens 
Oestrogens
Progestogens
The natural progestational hormone (progestogen) is progesterone (see Figs 34.2 and 34.3). This is secreted by the corpus luteum in the second part of the menstrual cycle, and by the placenta during pregnancy. Small amounts are also secreted by the testis and adrenal cortex.
Progestogens act, as do other steroid hormones, on nuclear receptors. The density of progesterone receptors is controlled by oestrogens (see above).
Preparations
There are two main groups of progestogens:
Actions
The pharmacological actions of the progestogens are in essence the same as the physiological actions of progesterone described above. Specific effects relevant to contraception are detailed below.
Pharmacokinetic aspects
Injected progesterone is bound to albumin, not to the sex steroid-binding globulin. Some is stored in adipose tissue. It is metabolised in the liver, and the products, pregnanolone and pregnanediol, are conjugated with glucuronic acid and excreted in the urine.
Unwanted effects
Unwanted effects of progestogens include weak androgenic actions. Other unwanted effects include acne, fluid retention, weight change, depression, change in libido, breast discomfort, premenstrual symptoms, irregular menstrual cycles and breakthrough bleeding. There is an increased incidence of thromboembolism.
Antiprogestogens
Mifepristone is a partial agonist at progesterone receptors. It sensitises the uterus to the action of prostaglandins. It is given orally and has a plasma half-life of 21 h. Mifepristone is used, in combination with a prostaglandin (e.g. gemeprost; see below), as a medical alternative to surgical termination of pregnancy (see clinical box).
Progestogens and antiprogestogens
Postmenopausal Hormone Replacement Therapy
At the menopause, whether natural or surgically induced, ovarian function decreases and oestrogen levels fall. There is a long history of disagreement regarding the pros and cons of hormone replacement therapy (HRT) in this context, with the prevailing wisdom undergoing several revisions over the years (see Davis et al., 2005). HRT normally involves the cyclic or continuous administration of low doses of one or more oestrogens, with or without a progestogen. Short-term HRT has some clear-cut benefits:
Oestrogen replacement does not reduce the risk of coronary heart disease, despite earlier hopes, nor is there evidence that it reduces age-related decline in cognitive function. Drawbacks include:
See Web links in the reference list for a useful table quantifying risks of cancer (breast, endometrium, ovary), venous thromboembolism, stroke and coronary artery disease in relation to age and duration of HRT use.
Oestrogens used in HRT can be given orally (conjugated estrogens, estradiol, estriol), vaginally (estriol), by transdermal patch (estradiol) or by subcutaneous implant (estradiol). Tibolone is marketed for the short-term treatment of symptoms of oestrogen deficiency. It has oestrogenic, progestogenic and weak androgenic activity, and can be used continuously without cyclical progesterone (avoiding the inconvenience of withdrawal bleeding).
Androgens
Testosterone is the main natural androgen. It is synthesised mainly by the interstitial cells of the testis, and in smaller amounts by the ovaries and adrenal cortex. Adrenal androgen production is controlled by adrenocorticotrophic hormone (ACTH, corticotrophin). As for other steroid hormones, cholesterol is the starting substance. Dehydroepiandrosterone and androstenedione are important intermediates. They are released from the gonads and the adrenal cortex, and converted to testosterone in the liver (see Fig. 34.3).
Actions
In general, the effects of exogenous androgens are the same as those of testosterone, and depend on the age and sex of the recipient. If given to prepubertal boys, the individuals concerned do not reach their full predicted height because of premature closure of the epiphyses of the long bones. In boys at the age of puberty, there is rapid development of secondary sexual characteristics (i.e. growth of facial, axillary and pubic hair, deepening of the voice), maturation of the reproductive organs and a marked increase in muscular strength. There is a growth spurt with an acceleration in the usual increase in height that occurs year on year in younger children, followed by cessation of linear growth. In adults, the anabolic effects can be accompanied by retention of salt and water. The skin thickens and may darken, and sebaceous glands become more active which can result in acne. Body weight and muscle mass increase, partly due to water retention. Androgens cause a feeling of well-being and an increase in physical vigour, and may increase libido. Whether they are responsible for sexual behaviour as such is controversial, as is their contribution to aggressive behaviour. Paradoxically, testosterone administration inhibits spermatogenesis, so reducing male fertility.
Administration of ‘male’ doses to women results in masculinisation, but lower doses (e.g. patches that release 300 µg of testosterone/day) restore plasma testosterone to normal female concentrations and improve sexual dysfunction in women following ovariectomy, without adverse effects (Shifren et al., 2000; Braunstein et al., 2005).
Mechanism of action
In most target cells, testosterone works through an active metabolite, dihydrotestosterone, to which it is converted locally by a 5α-reductase enzyme. In contrast, testosterone itself causes virilisation of the genital tract in the male embryo and regulates LH/ICSH production in anterior pituitary cells. Testosterone and dihydrotestosterone modify gene transcription by interacting with nuclear receptors.
Preparations
Testosterone itself can be given by subcutaneous implantation or by transdermal patches (male replacement dose approximately 2.5 mg/day. Various esters (e.g. enanthate and proprionate) are given by intramuscular depot injection. Testosterone undecanoate and mesterolone can be given orally.
Pharmacokinetic aspects
If given orally, testosterone is rapidly metabolised in the liver. Virtually all testosterone in the circulation is bound to plasma protein—mainly to the sex steroid-binding globulin. The elimination half-life of free testosterone is short (10–20 min). It is inactivated in the liver by conversion to androstenedione (see Fig. 34.3). This has weak androgenic activity in its own right and can be reconverted to testosterone, although approximately 90% of testosterone is eliminated as metabolites rather than the parent compound. Synthetic androgens are less rapidly metabolised, and some are excreted in the urine unchanged.
Unwanted effects
Unwanted effects of androgens include eventual decrease of gonadotrophin release, with resultant infertility, and salt and water retention leading to oedema. Adenocarcinoma of the liver has been reported. Androgens impair growth in children (via premature fusion of epiphyses), cause acne and lead to masculinisation in girls. Adverse effects of testosterone replacement and monitoring for these are reviewed by Rhoden & Morgentaler (2004).
The clinical uses of androgens are given in the clinical box.
Androgens and the hormonal control of the male reproductive system
Anabolic Steroids
Androgens can be modified chemically to alter the balance of anabolic and other effects. Such ‘anabolic steroids’ (e.g. nandrolone) increase protein synthesis and muscle development, but clinical use (e.g. in debilitating disease) has been disappointing. They are used in the therapy of aplastic anaemia and (notoriously) abused by some athletes (Ch. 58), as is testosterone itself. Unwanted effects are described above, under Androgens. In addition, cholestatic jaundice, liver tumours and increased risk of coronary heart disease are recognised adverse effects of high-dose anabolic steroids.
Antiandrogens
Both oestrogens and progestogens have antiandrogen activity, oestrogens mainly by inhibiting gonadotrophin secretion and progestogens by competing at androgen receptors in target organs. Cyproterone is a derivative of progesterone and has weak progestational activity. It is a partial agonist at androgen receptors, competing with dihydrotestosterone for receptors in androgen-sensitive target tissues. Through its effect in the hypothalamus, it depresses the synthesis of gonadotrophins. It is used as an adjunct in the treatment of prostatic cancer during initiation of GnRH treatment (see below). It is also used in the therapy of precocious puberty in males, and of masculinisation and acne in women. It also has a central nervous system effect, decreasing libido, and has been used to treat hypersexuality in male sexual offenders.2
Flutamide is a non-steroidal antiandrogen used with GnRH in the treatment of prostate cancer.
Drugs can have antiandrogen action by inhibiting synthetic enzymes. Finasteride inhibits the enzyme (5α-reductase) that converts testosterone to dihydrotestosterone (Fig. 34.3). This which has greater affinity than testosterone for androgen receptors in the prostate gland. Finasteride is well absorbed after oral administration, has a half-life of about 7 h, and is excreted in the urine and faeces. It is used to treat benign prostatic hyperplasia, although α1-adrenoceptor antagonists, for example terazosin or tamsulosin (Ch. 14), are more effective (working by the entirely different mechanism of relaxing smooth muscle in the capsule of the prostate gland and opposing α1-adrenoceptor-mediated prostatic growth). Surgery is the preferred option (especially by surgeons).
Gonadotrophin-Releasing Hormone: Agonists and Antagonists
Gonadotrophin-releasing hormone is a decapeptide that controls the secretion of FSH and LH by the anterior pituitary. Secretion of GnRH is controlled by neural input from other parts of the brain, and through negative feedback by the sex steroids (Figs 34.1 and 34.5). Exogenous androgens, oestrogens and progestogens all inhibit GnRH secretion, but only progestogens exert this effect at doses that do not have marked hormonal actions on peripheral tissues, presumably because progesterone receptors in the reproductive tract are sparse unless they have been induced by previous exposure to oestrogen. Danazol (see below) is a synthetic steroid that inhibits release of GnRH and, consequently, of gonadotrophins (FSH and LH). Clomiphene is an oestrogen antagonist that stimulates gonadotrophin release by inhibiting the negative feedback effects of endogenous oestrogen; it is used to treat infertility (see above and Fig. 34.5).
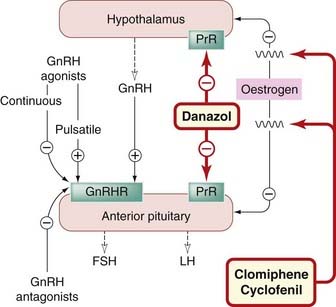
Fig. 34.5 Regulation of gonadotrophin (follicle-stimulating hormone, FSH; luteinising hormone, LH) release from the anterior pituitary.
Synthetic GnRH is termed gonadorelin. Numerous analogues of GnRH, both agonists and antagonists, have been synthesised. Buserelin, leuprorelin, goserelin and nafarelin are agonists, the last being 200 times more potent than endogenous GnRH.
Pharmacokinetics and clinical use
Gonadotrophin-releasing hormone agonists, given by subcutaneous infusion in pulses to mimic physiological secretion of GnRH, stimulate gonadotrophin release (Fig. 34.5) and induce ovulation. They are absorbed intact following nasal administration (Ch. 8). Continuous use, by nasal spray or as depot preparations, stimulates gonadotrophin release transiently, but then paradoxically inhibits gonadotrophin release (Fig. 34.5) because of downregulation (desensitisation) of GnRH receptors in the pituitary. GnRH analogues are given in this fashion to cause gonadal suppression in various sex hormone-dependent conditions, including prostate and breast cancers, endometriosis (endometrial tissue outside the uterine cavity) and large uterine fibroids. Continuous, non-pulsatile administration inhibits spermatogenesis and ovulation, raising the possibility (which is under investigation) that GnRH analogues could be useful as contraceptives. GnRH agonists are used by specialists in infertility treatment, not to stimulate ovulation (which is achieved using gonadotrophin preparations) but to suppress the pituitary before administration of FSH or HCG (see below). It was originally hoped that GnRH antagonists would be useful for contraception, but this has not been realised.
Unwanted effects of GnRH analogues
Unwanted effects of GnRH agonists in women, for example flushing, vaginal dryness and bone loss, result from hypo-oestrogenism. The initial stimulation of gonadotrophin secretion on starting treatment can cause transient worsening of pain from bone metastases in men with prostate cancer, so treatment is started only after the patient has received an androgen receptor antagonist such as flutamide (see above and Ch. 55).
Danazol
Actions and pharmacokinetics
Danazol inhibits gonadotrophin secretion (especially the mid-cycle surge), and consequently reduces oestrogen synthesis in the ovary (Fig. 34.5). In men, it reduces androgen synthesis and spermatogenesis. It has androgenic activity. It is orally active and metabolised in the liver.
Danazol is used in sex hormone-dependent conditions including endometriosis, breast dysplasia and gynaecomastia. An additional special use is to reduce attacks of swelling in hereditary angio-oedema (Ch. 27).
Unwanted effects are common, and include gastrointestinal disturbances, weight gain, fluid retention, dizziness, menopausal symptoms, muscle cramps and headache. Danazol is virilising in women.
Gonadotrophins and Analogues
Gonadotrophins (FSH, LH and HCG) are glycoproteins produced and secreted by the anterior pituitary (see Ch. 32) or chorion and placenta. Large amounts of gonadotrophins are present in the urine of women following the menopause, in whom oestrogen no longer exerts feedback inhibition on the pituitary, which consequently secretes large amounts of FSH and LH.3 The chorion and placenta secrete HCG.
Preparations
Gonadotrophins are extracted from urine of pregnant (HCG) or postmenopausal women (human menopausal gonadotrophin, which contains a mixture of FSH and LH). Recombinant FSH (follitropin) and LH (lutropin) are also available.
Pharmacokinetics and clinical use
Gonadotrophin preparations are given by injection. They are used to treat infertility caused by lack of ovulation as a result of hypopituitarism, or following failure of treatment with clomiphene; they are also used by specialists to induce ovulation to enable eggs to be collected for in vitro fertilisation. For this use, gonadotrophin is usually administered after secretion of endogenous FSH and LH has been suppressed (see above). Gonadotrophins are also sometimes used in men with infertility caused by a low sperm count as a result of hypogonadotrophic hypogonadism (a disorder that is sometimes accompanied by lifelong anosmia, i.e. lack of sense of smell). (Gonadotrophins do not, of course, work for patients whose low sperm count is the result of primary testicular failure.) HCG has been used to stimulate testosterone synthesis in boys with delayed puberty, but testosterone is usually preferred.
Gonadotrophin-releasing hormone and gonadotrophins
Drugs Used for Contraception
Oral Contraceptives
There are two main types of oral contraceptives:
The Combined Pill
The combined oral contraceptive pill is extremely effective, at least in the absence of intercurrent illness and of treatment with potentially interacting drugs (see below). The oestrogen in most combined preparations (second-generation pills)4 is ethinylestradiol, although a few preparations contain mestranol instead. The progestogen may be norethisterone, levonorgestrel, ethynodiol, or—in ‘third-generation’ pills—desogestrel or gestodene, which are more potent, have less androgenic action and cause less change in lipoprotein metabolism, but which probably cause a greater risk of thromboembolism than do second-generation preparations. The oestrogen content is generally 20–50 µg of ethinylestradiol or its equivalent, and a preparation is chosen with the lowest oestrogen and progestogen content that is well tolerated and gives good cycle control in the individual woman. This combined pill is taken for 21 consecutive days followed by 7 pill-free days, which causes a withdrawal bleed. Normal cycles of menstruation usually commence fairly soon after discontinuing treatment, and permanent loss of fertility (which may be a result of early menopause rather than a long-term consequence of the contraceptive pill) is rare.
The mode of action is as follows:
They may also interfere with the coordinated contractions of the cervix, uterus and fallopian tubes that facilitate fertilisation and implantation.
Hundreds of millions of women worldwide have used this method since the 1960s, and in general the combined pill constitutes a safe and effective method of contraception. There are distinct health benefits from taking the pill (see below), and serious adverse effects are rare. However, minor unwanted effects constitute drawbacks to its use, and several important questions need to be considered.
Questions that need to be considered
Is there an increased risk of cardiovascular disease (venous thromboembolism, myocardial infarction, stroke)?
With second-generation pills (oestrogen content less than 50 µg), the risk of thromboembolism is small (incidence approximately 15 per 100 000 users per year, compared with 5 per 100 000 non-pregnant non-users per year or 60 episodes of thromboembolism per 100 000 pregnancies). The risk is greatest in subgroups with additional factors, such as smoking (which increases risk substantially) and long-continued use of the pill, especially in women over 35 years of age. For preparations containing the third-generation progestogens desogestrel or gestodene, the incidence of thromboembolic disease is approximately 25 per 100 000 users per year, which is still a small absolute risk compared with the risk of thromboemolism in an unwanted pregnancy. In general, provided risk factors, e.g. smoking, hypertension and obesity, have been identified, combined oral contraceptives are safe for most women for most of their reproductive lives.
Is blood pressure increased?
A marked increase in arterial blood pressure occurs in a small percentage of women shortly after starting the combined oral contraceptive pill. This is associated with increased circulating angiotensinogen, and disappears when treatment is stopped. Blood pressure is therefore monitored carefully when oral contraceptive treatment is started, and an alternative contraceptive substituted if necessary.
Beneficial effects
The combined pill markedly decreases menstrual symptoms such as irregular periods and intermenstrual bleeding. Iron deficiency anaemia and premenstrual tension are reduced, as are benign breast disease, uterine fibroids and functional cysts of the ovaries. Unwanted pregnancy, carrying a maternal mortality ranging from 1 in 10 000 in developed countries to 1 in 150 in Africa, is avoided.
The Progestogen-Only Pill
The drugs used in progestogen-only pills include norethisterone, levonorgestrel or ethynodiol. The pill is taken daily without interruption. The mode of action is primarily on the cervical mucus, which is made inhospitable to sperm. The progestogen probably also hinders implantation through its effect on the endometrium and on the motility and secretions of the fallopian tubes (see above).
Potential beneficial and unwanted effects
Progestogen-only contraceptives offer a suitable alternative to the combined pill for some women in whom oestrogen is contraindicated, and are suitable for women whose blood pressure increases unacceptably during treatment with oestrogen. However, their contraceptive effect is less reliable than that of the combination pill, and missing a dose may result in conception. Disturbances of menstruation (especially irregular bleeding) are common. Only a small proportion of women use this form of contraception, so long-term safety data are less reliable than for the combined pill.
Pharmacokinetics of oral contraceptives: drug interactions
Combined and progestogen-only oral contraceptives are metabolised by hepatic cytochrome P450 enzymes. Because the minimum effective dose of oestrogen is used (in order to avoid excess risk of thromboembolism), any increase in its clearance may result in contraceptive failure, and indeed enzyme-inducing drugs can have this effect not only for combined but also for progesterone-only pills. Such drugs include (par excellence) rifampicin and rifabutin, as well as carbamazepine, phenytoin and others. Enterohepatic recycling of oestrogen is mentioned above. Broad-spectrum antibiotics such as amoxicillin can disturb this by altering the intestinal flora, and cause failure of the combined pill. This does not occur with progestogen-only pills.
Oral contraceptives
The combined pill
Other Drug Regimens Used for Contraception
Postcoital (Emergency) Contraception
Oral administration of levonorgestrel, alone or combined with oestrogen, is effective if taken within 72 h of unprotected intercourse and repeated 12 h later. Nausea and vomiting are common (and the pills may then be lost: replacement tablets can be taken with an antiemetic such as domperidone). Insertion of an intrauterine device is more effective than hormonal methods, and works up to 5 days after intercourse.
Long-Acting Progestogen-Only Contraception
Medroxyprogesterone can be given intramuscularly as a contraceptive. This is effective and safe. However, menstrual irregularities are common, and infertility may persist for many months after cessation of treatment.
Levonorgestrel implanted subcutaneously in non-biodegradable capsules is used by approximately 3 million women worldwide. This route of administration avoids first-pass metabolism. The capsules release their progestogen content slowly over 5 years. Irregular bleeding and headache are common.
A levonorgestrel-impregnated intrauterine device has contraceptive action for 35 years.
The Uterus
The physiological and pharmacological responses of the uterus vary at different stages of the menstrual cycle and during pregnancy.
The Motility of the Uterus
Uterine muscle contracts rhythmically both in vitro and in vivo, contractions originating in the muscle itself. Myometrial cells in the fundus act as pacemakers and give rise to conducted action potentials. The electrophysiological activity of these pacemaker cells is regulated by the sex hormones.
The non-pregnant human uterus contracts spontaneously but weakly during the first part of the cycle, and more strongly during the luteal phase and during menstruation. Uterine movements are depressed in early pregnancy because oestrogen, potentiated by progesterone, hyperpolarises myometrial cells. This suppresses spontaneous contractions. Towards the end of gestation, however, contractions recommence; these increase in force and frequency, and become fully coordinated during parturition. The nerve supply to the uterus includes both excitatory and inhibitory sympathetic components: adrenaline, acting on β2 adrenoceptors, inhibits uterine contraction, whereas noradrenaline, acting on α-adrenoceptors, stimulates contraction.
Drugs that Stimulate the Uterus
Drugs that stimulate the pregnant uterus and are important in obstetrics include oxytocin, ergometrine and prostaglandins.
Oxytocin
As explained in Chapter 32, the neurohypophyseal hormone oxytocin (an octapeptide) regulates myometrial activity. Oxytocin release is stimulated by cervical dilatation, and by suckling, but its role in parturition is incompletely understood.
Oxytocin contracts the uterus. Oestrogen induces oxytocin receptor synthesis and, consequently, the uterus at term is highly sensitive to this hormone. Given by slow intravenous infusion to induce labour, oxytocin causes regular coordinated contractions that travel from fundus to cervix. Both amplitude and frequency of these contractions are related to dose, the uterus relaxing completely between contractions during low-dose infusion. Larger doses further increase the frequency of the contractions, and there is incomplete relaxation between them. Still higher doses cause sustained contractions that interfere with blood flow through the placenta and cause fetal distress or death.
Oxytocin contracts myoepithelial cells in the mammary gland, which causes ‘milk let-down’—the expression of milk from the alveoli and ducts. It also has a vasodilator action. A weak antidiuretic action can result in water retention, which can be problematic in patients with cardiac or renal disease, or with pre-eclampsia.5 Oxytocin and oxytocin receptors are also found in the brain, particularly in the limbic system, and are believed to play a role in mating and parenting behaviour.
The clinical use of synthetic oxytocin is given in the box.
Oxytocin can be given by intravenous injection or intramuscularly, but is most often given by intravenous infusion. It is inactivated in the liver and kidneys, and by circulating placental oxytocinase.
Unwanted effects of oxytocin include dose-related hypotension, due to vasodilatation, with associated reflex tachycardia. Its antidiuretic hormone-like effect on water excretion by the kidney causes water retention and, unless water intake is curtailed, consequent hyponatraemia.
Ergometrine
Ergot (Claviceps purpurea) is a fungus that grows on rye and contains a surprising variety of pharmacologically active substances (see Ch. 15). Ergot poisoning, which was once common, was often associated with abortion. In 1935, ergometrine was isolated and was recognised as the oxytocic principle in ergot.
Ergometrine contracts the human uterus. This action depends partly on the contractile state of the organ. On a contracted uterus (the normal state following delivery), ergometrine has relatively little effect. However, if the uterus is inappropriately relaxed, ergometrine initiates strong contraction, thus reducing bleeding from the placental bed (the raw surface from which the placenta has detached). Ergometrine also has a moderate vasoconstrictor action.
The mechanism of action of ergometrine on smooth muscle is not understood. It is possible that it acts partly on α-adrenoceptors, like the related alkaloid ergotamine (see Ch. 14), and partly on 5-hydroxytryptamine receptors.
The clinical use of ergometrine is given in the box.
Ergometrine can be given orally, intramuscularly or intravenously. It has a very rapid onset of action and its effect lasts for 3–6 h.
Ergometrine can produce vomiting, probably by an effect on dopamine D2 receptors in the chemoreceptor trigger zone (see Fig. 29.5). Vasoconstriction with an increase in blood pressure associated with nausea, blurred vision and headache can occur, as can vasospasm of the coronary arteries, resulting in angina.
Prostaglandins
Prostaglandins are discussed in detail in Chapter 17. The endometrium and myometrium have substantial prostaglandin-synthesising capacity, particularly in the second, proliferative phase of the menstrual cycle. Prostaglandin (PG)F2α is generated in large amounts, and has been implicated in the ischaemic necrosis of the endometrium that precedes menstruation (although it has relatively little vasoconstrictor action on many human blood vessels, in contrast to some other mammalian species). Vasodilator prostaglandins, PGE2 and PGI2 (prostacyclin), are also generated by the uterus.
In addition to their vasoactive properties, the E and F prostaglandins contract the non-pregnant as well as the pregnant uterus. The sensitivity of uterine muscle to prostaglandins increases during gestation. Their role in parturition is not fully understood, but as cyclo-oxygenase inhibitors can delay labour (see below), they probably play some part in this.
Prostaglandins also play a part in two of the main disorders of menstruation: dysmenorrhoea (painful menstruation) and menorrhagia (excessive blood loss). Dysmenorrhoea is associated with increased production of PGE2 and PGF2α; non-steroidal anti-inflammatory drugs, which inhibit prostaglandin biosynthesis (see Ch. 26), are used to treat dysmenorrhoea. Menorrhagia, in the absence of uterine pathology, may be caused by a combination of increased vasodilatation and reduced haemostasis. Increased generation by the uterus of PGI2 (which inhibits platelet aggregation) could impair haemostasis as well as causing vasodilatation. Non-steroidal anti-inflammatory drugs (e.g. mefenamic acid) are used to treat menorrhagia as well as dysmenorrhoea.
Prostaglandin preparations
Prostaglandins of the E and F series promote coordinated contractions of the body of the pregnant uterus, while relaxing the cervix. E and F prostaglandins reliably cause abortion in early and middle pregnancy, unlike oxytocin which generally does not cause expulsion of the uterine contents at this stage. The prostaglandins used in obstetrics are dinoprostone (PGE2), carboprost (15-methyl PGF2α) and gemeprost or misoprostol (PGE1 analogues). Dinoprostone can be given intravaginally as a gel or as tablets, or by the extra-amniotic route as a solution. Carboprost is given by deep intramuscular injection. Gemeprost or misoprostol are given intravaginally.
Unwanted effects
Unwanted effects include uterine pain, nausea and vomiting, which occur in about 50% of patients when the drugs are used as abortifacients. Dinoprost may cause cardiovascular collapse if it escapes into the circulation after intra-amniotic injection. Phlebitis can occur at the site of intravenous infusion. When combined with mifepristone, a progestogen antagonist that sensitises the uterus to prostaglandins, lower doses of the prostaglandins (e.g. misoprostol) can be used to terminate pregnancy and side effects are reduced.
See the clinical box for the clinical uses of prostaglandins.
Clinical uses of drugs acting on the uterus
Myometrial stimulants (oxytocics)
Drugs that Inhibit Uterine Contraction
Selective β2-adrenoceptor agonists, such as ritodrine or salbutamol, inhibit spontaneous or oxytocin-induced contractions of the pregnant uterus. These uterine relaxants are used in selected patients to prevent premature labour occurring between 22 and 33 weeks of gestation in otherwise uncomplicated pregnancies. They can delay delivery by 48 h, time that can be used to administer glucocorticoid therapy to the mother so as to mature the lungs of the baby and reduce neonatal respiratory distress. It has been difficult to demonstrate that any of the drugs used to delay labour improve the outcome for the baby. Risks to the mother, especially pulmonary oedema, increase after 48 h, and myometrial response is reduced, so prolonged treatment is avoided. Cyclo-oxygenase inhibitors (e.g. indometacin) inhibit labour, but their use could cause problems in the baby, including renal dysfunction and delayed closure of the ductus arteriosus, both of which are influenced by endogenous prostaglandins.
An oxytocin receptor antagonist, atosiban, provides an alternative to a β2-adrenoceptor agonist. It is given as an intravenous bolus followed by an intravenous infusion for not more than 48 h. Adverse effects include vasodilatation, nausea, vomiting and hyperglycaemia.
Drugs acting on the uterus
Erectile Dysfunction
Erectile function depends on complex interactions between physiological and psychological factors. Erection is caused by vasorelaxation in the arteries and arterioles supplying the erectile tissue. This increases penile blood flow; the consequent increase in sinusoidal filling compresses the venules, occluding venous outflow and causing erection. During sexual intercourse, reflex contraction of the ischiocavernosus muscles compresses the base of the corpora cavernosa, and the intracavernosal pressure can reach several hundred millimetres of mercury during this phase of rigid erection. Innervation of the penis includes autonomic and somatic nerves. Nitric oxide is probably the main mediator of erection and is released both from nitrergic nerves and from endothelium (Ch. 20; Fig. 20.6).
Erectile function is adversely affected by several therapeutic drugs (including many antipsychotic, antidepressant and antihypertensive agents), and psychiatric and vascular disease (especially if this has caused endothelial dysfunction) can themselves cause erectile dysfunction, which is common in middle-aged and older men, even if they have no psychiatric or cardiovascular problems.6 There are several organic causes, including hypogonadism (see above), hyperprolactinaemia (see Ch. 32), arterial disease and various causes of neuropathy (most commonly diabetes), but often no organic cause is identified.
Over the centuries, there has been a huge trade in parts of various creatures that have the misfortune to bear some fancied resemblance to human genitalia, in the pathetic belief that consuming these will restore virility or act as an aphrodisiac (i.e. a drug that stimulates libido). Alcohol (Ch. 48) ‘provokes the desire but takes away the performance’, and cannabis (Ch. 18) can also release inhibitions and probably does the same. Yohimbine (an α2 adrenoceptor antagonist; Ch. 14) may have some positive effect in this regard, but trials have proved inconclusive. Apomorphine (a dopamine agonist; Ch. 38) causes erections in humans as well as in rodents when injected subcutaneously, but it is a powerful emetic, an effect that is usually regarded as socially unacceptable in this context. Despite this rather obvious disadvantage, a sublingual preparation is licensed for erectile dysfunction.7 Nausea is said to subside with continued use—but really!
The generally negative picture picked up somewhat when it was found that injecting vasodilator drugs directly into the corpora cavernosa causes penile erection. Papaverine (Ch. 22), if necessary with the addition of phentolamine, was used in this way. The route of administration is not acceptable to most men, but diabetics in particular are often not needle-shy, and this approach was a real boon to many such patients. PGE1 (alprostadil) is often combined with other vasodilators when given intracavernosally. It can also be given transurethrally as an alternative (albeit still a somewhat unromantic one) to injection. Adverse effects of all these drugs include priapism, which is no joke. Treatment consists of aspiration of blood (using sterile technique) and, if necessary, cautious intracavernosal administration of a vasoconstrictor such as phenylephrine. Intracavernosal and transurethral preparations are still available, but orally active phosphodiesterase inhibitors are now generally the drugs of choice.
Phosphodiesterase Type V Inhibitors
Sildenafil, the first selective phosphodiesterase type V inhibitor (see also Chs 20 and 22), was found accidentally to influence erectile function.8 Tadalafil and vardenafil are also phosphodiesterase type V inhibitors licensed to treat erectile dysfunction. Tadalafil is longer acting than sildenafil. In contrast to intracavernosal vasodilators, phosphodiesterase type V inhibitors do not cause erection independent of sexual desire, but enhance the erectile response to sexual stimulation. They have transformed the treatment of erectile dysfunction.
Mechanism of action
Phosphodiesterase V is the isoform that inactivates cGMP. Nitrergic nerves release nitric oxide (or a related nitrosothiol) which diffuses into smooth muscle cells, where it activates guanylyl cyclase. The resulting increase in cytoplasmic cGMP mediates vasodilatation via activation of protein kinase G (Fig 4.10). Consequently, inhibition of phosphodiesterase V potentiates the effect on penile vascular smooth muscle of endothelium-derived nitric oxide and of nitrergic nerves that are activated by sexual stimulation (Fig. 34.6). Other vascular beds are also affected, suggesting other possible uses, notably in pulmonary hypertension (Ch. 22).
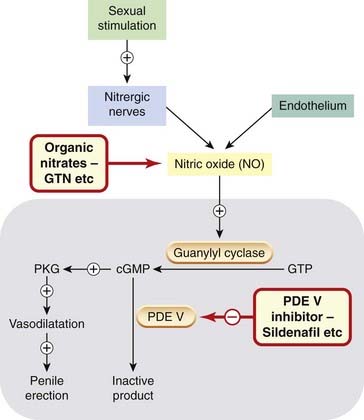
Fig. 34.6 Mechanism of phosphodiesterase V (PDE V) inhibitors on penile erection, and of the interaction of PDE V inhibitors with organic nitrates.
The large grey rectangle denotes a vascular smooth muscle cell in the corpora cavernosa. Sexual stimulation releases nitric oxide (NO) from nitrergic nerves and this activates guanylyl cyclase, increasing cGMP production and hence activating protein kinase G (PKG), causing vasodilatation and penile erection. cGMP is inactivated by PDE V, so PDE V inhibitors (e.g. sildenafil) potentiate NO and promote penile erection. NO from organic nitrates such as glyceryl trinitrate (GTN) is also potentiated leading to generalised vasodilatation and hypotension.
Pharmacokinetic aspects and drug interactions
Peak plasma concentrations of sildenafil occur approximately 30–120 min after an oral dose and are delayed by eating, so it is taken an hour or more before sexual activity. It is given as a single dose as needed. It is metabolised by CYP3A4, which is induced by carbamazepine, rifampicin and barbiturates, and inhibited by cimetidine, macrolide antibiotics, antifungal imidazolines, some antiviral drugs (such as ritonavir) and also by grapefruit juice (Ch. 56). These drugs can potentially interact with sildenafil in consequence. Tadalafil has a longer half-life than sildenafil, so can be taken longer before sexual activity. A clinically important pharmacodynamic interaction of all phosphodiesterase V inhibitors occurs with all organic nitrates,9 which work through increasing cGMP (Ch. 20) and are therefore markedly potentiated by sildenafil (Fig. 34.6). Consequently, concurrent nitrate use, including use of nicorandil, contraindicates the use of any phosphodiesterase type V inhibitor.
Unwanted effects
Many of the unwanted effects of phosphodiesterase type V inhibitors are caused by vasodilatation in other vascular beds; these effects include hypotension, flushing and headache. Visual disturbances have occasionally been reported and are of concern because sildenafil has some action on phosphodiesterase VI, which is present in the retina and important in vision. The manufacturers advise that sildenafil should not be used in patients with hereditary retinal degenerative diseases (such as retinitis pigmentosa) because of the theoretical risk posed by this. Vardenafil is more selective for the type V isozyme than is sildenafil (reviewed by Doggrell, 2005), but is also contraindicated in patients with hereditary retinal disorders.
References and Further Reading
Sex hormones and their control
Barrett-Connor E., Mosca L., Collins P., et al. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N. Eng. J. Med.. 2006;355:125-137. (Reduced breast cancer)
Chen Z., Yuhanna I.S., Galcheva-Gargova Z., et al. Estrogen receptor-alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J. Clin. Invest. 1999;103:401-406. (Acute vasodilator action of oestrogen may involve membrane ER rather than the classic intracellular receptor pathway)
Gruber C.J., Tschugguel W., Schneeberger C., Huber J.C. Production and actions of estrogens. N. Engl. J. Med.. 2002;346:340-352. (Review focusing on the new biochemical aspects of the action of oestrogen—including phyto-oestrogens and selective oestrogen receptor modulators—as well as physiological and clinical aspects)
Huirne J.A.F., Lambalk C.B. Gonadotrophin-releasing hormone receptor antagonists. Lancet. 2001;358:1793-1803. (Review discussing their clinical potential)
Olive D.L., Pritts E.A. Treatment of endometriosis. N. Engl. J. Med.. 2001;34:266-275. (Critical review of existing evidence—which is thin—forms the basis for sensible recommendations regarding treatment of pelvic pain or infertility from endometriosis using oral contraceptives and GnRH agonist therapy with oestrogen–progestin add-back)
Rhoden E.L., Morgentaler A. Risks of testosterone-replacement therapy and recommendations for monitoring. N. Engl. J. Med.. 2004;350:482-492. (Review)
Vogel V., Constantino J., Wickerman L., et al. Effects of tamoxifen vs. raloxifene on the risk of developing invasive breast cancer and other disease outcomes. JAMA. 2006;295:2727-2741. (Raloxifene had similar efficacy as tamoxifen with fewer thrombotic events)
Djerassi C. This man’s pill: reflections on the 50th birthday of the pill. New York: Oxford University Press; 2001. (Scientific and autobiographical memoir by polymath steroid chemist who worked on ‘the pill’ at its inception under Syntex in Mexico, and has continued thinking about human reproduction in a broad biological and biosocial sense ever since)
Braunstein G.D., Sundwall D.A., Katz M., et al Safety and efficacy of a testosterone patch for the treatment of hypoactive sexual desire disorder in surgically menopausal women: a randomized, placebo-controlled trial 165. 2005 Arch. Intern. Med.
Davis S.R., Dinatale I., Rivera-Woll L., Davison S. Postmenopausal hormone therapy: from monkey glands to transdermal patches. J. Endocrinol.. 2005;185:207-222. (Reviews the history of knowledge of the menopause and the development of hormonal therapy for climacteric complaints, and summarises current evidence for specific benefits and risks of hormone treatment)
Hulley S., Grady D., Bush T., et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. JAMA. 1998;280:605-613. (A total of 2763 postmenopausal women who had suffered a previous coronary event randomised to active or placebo and observed for a mean of 4.1 years. The incidence of fatal myocardial infarction was similar in the two groups, despite favourable changes in low- and high-density lipoproteins cholesterol in the HRT group. Venous thromboembolism was increased by a factor of 2.89 in the active group)
Shifren J.L., Braunstein G.D., Simon J.A., et al. Transdermal testosterone treatment in women with impaired sexual function after oophorectomy. N. Engl. J. Med.. 2000;343:682-688. (Transdermal testosterone improves sexual function and psychological well-being in women who have undergone oophorectomy and hysterectomy)
Norwitz E.R., Robinson J.N., Challis J.R. The control of labor. N. Engl. J. Med.. 1999;341:660-666. (Review)
Thornton S., Vatish M., Slater D. Oxytocin antagonists: clinical and scientific considerations. Exp. Physiol.. 2001;86:297-302. (Reviews rationale for uterine relaxants in preterm labour; evidence for administering atosiban; and the role of oxytocin, vasopressin and their receptors in the onset of labour)
Andersson K.-E. Pharmacology of penile erection. Pharmacol. Rev.. 2001;53:417-450. (Scholarly review covering central and peripheral regulation, and a very wide-ranging coverage of the pharmacology)
Doggrell S.A. Comparison of clinical trials with sildenafil, vardenafil and tadalafil in erectile dysfunction. Expert Opin. Pharmacother.. 2005;6:75-84. (Vardenafil is similarly effective to sildenafil. Its only advantage is that it does not inhibit phosphodiesterase VI to alter colour perception, a rare side effect that sometimes occurs with sildenafil. Tadalafil has a longer duration of action)
http://www.mhra.gov.uk/mhra/drugsafetyupdate (A useful table quantifying risks of cancer [breast, endometrium, ovary], venous thromboembolism, stroke and coronary artery disease in relation to age and duration of HRT use)
1Recognition that negative feedback is central to endocrine control was a profound insight, made in 1930 by Dorothy Price, a laboratory assistant in the University of Chicago experimenting on effects of testosterone in rats. She referred to it as ‘reciprocal influence’.
2Very different doses are used for these different conditions, for example 2 mg/day for acne, 100 mg/day for hypersexuality and 300 mg/day for prostatic cancer.
3This forms the basis for the standard blood test, estimation of plasma LH/FSH concentrations, to confirm whether a woman is postmenopausal.
4The first-generation pills, containing more than 50 µg of oestrogen, were shown in the 1970s to be associated with an increased risk of deep vein thrombosis and pulmonary embolism.
5Eclampsia is a pathological condition (involving, among other things, high blood pressure, swelling and seizures) that occurs in pregnant women.
6In randomised controlled trials, an appreciable proportion of men who discontinued treatment because of erectile dysfunction had been receiving placebo.
7Ironically so, because apomorphine was used as ‘aversion therapy’ in a misguided attempt to ‘cure’ homosexuality by conditioning individuals to associate homoerotic stimuli with nausea and vomiting, during the not-so-very-far-off time when homosexuality was classified as a psychiatric disease (‘only apomorphine cures’—William Burroughs, Naked Lunch. Grove Press, 1966).
8Sildenafil was originally intended to treat angina, but volunteers in early phase trials reported an effect on affairs of the heart in a quite different anatomical region from the precordium.
9This is important not only for sufferers from angina who take nitrates such as glyceryl trinitrate or isosorbide mononitrate therapeutically or prophylactically and are at risk of hypotension because of coronary artery disease, but also asymptomatic individuals who take amyl nitrate recreationally (‘poppers’) because of its effect on pelvic musculature.