28 The kidney
Overview
We set the scene with a brief outline of renal physiology based on the functional unit of the kidney—the nephron—before describing drugs that affect renal function. Subsequent emphasis is on diuretics—drugs that increase the excretion of Na+ ions and water. We also consider briefly other drugs that are used in treatment of patients with renal failure and urinary tract disorders.
Introduction
The main drugs that work by altering renal function—the diuretics—are crucial for the management of cardiovascular disease (Chs 21 and 22) as well as patients with renal disease. The kidneys are the main organ by which drugs and their metabolites are eliminated from the body (Ch. 9), and so in renal impairment dosing regimens of many drugs must be adapted. Furthermore, the kidneys are a target for various kinds of drug toxicity (Ch. 57). Antihypertensive drugs (commonly indicated in kidney disease) are covered in Chapter 22, immunosuppressant drugs (effective in several of the diseases that can cause renal failure, and crucial for maintaining the health of patients who have received a kidney transplant) in Chapter 26 and antibacterial drugs (used to treat renal and urinary tract infections) in Chapter 50. Patients with anaemia due to chronic renal failure benefit greatly from epoetin (Ch. 25). In the present chapter we focus on the main drugs that act on the renal tubules, namely diuretics—drugs that increase the excretion of Na+ ions and water. We also consider briefly other drugs that are used in treating renal failure (concentrating on acid–base and electrolyte aspects) and urinary tract disorders.
Outline of Renal Function
The main function of the kidney is to maintain the constancy of the ‘interior environment’ by eliminating waste products and by regulating the volume, electrolyte content and pH of the extracellular fluid in the face of varying dietary intake and other environmental (e.g. climatic) demands.
The kidneys receive about a quarter of the cardiac output. From the several hundred litres of plasma that flow through them each day, they filter (in a 70 kg human) approximately 120 litres per day, 11 times the total extracellular fluid volume. This filtrate is similar to plasma apart from the absence of protein. As it passes through the renal tubule, about 99% of the filtered water, and much of the filtered Na+, is reabsorbed, and some substances are secreted into it from the blood. Eventually, approximately 1.5 litres is voided as urine per 24 h under usual conditions (Table 28.1).
Each kidney consists of an outer cortex, an inner medulla and a hollow pelvis, which empties into the ureter. The functional unit is the nephron, of which there are approximately 1.4 × 106 in each kidney (approximately half this number in people with hypertension), with considerable variation between individuals and an age-related decline.
The Structure and Function of the Nephron
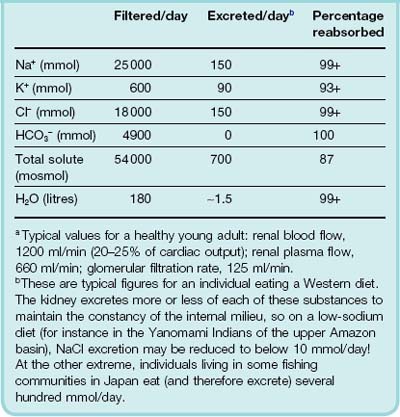
Each nephron consists of a glomerulus, proximal tubule, loop of Henle, distal convoluted tubule and collecting duct—Figure 28.1. The glomerulus comprises a tuft of capillaries projecting into a dilated end of the renal tubule. Most nephrons lie largely or entirely in the cortex. The remaining 12%, called the juxtamedullary nephrons, have their glomeruli and convoluted tubules next to the junction of the medulla and cortex, and their loops of Henle pass deep into the medulla.
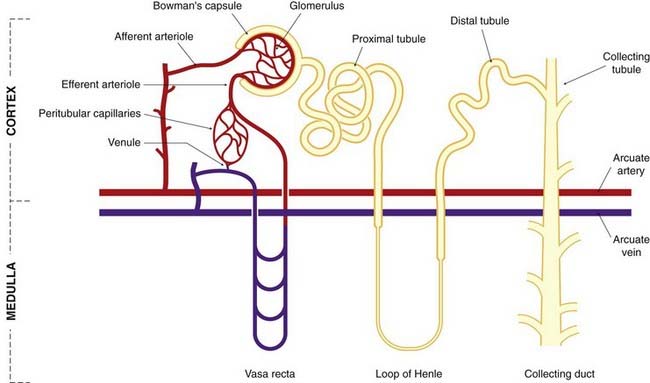
Fig. 28.1 Simplified diagram of a juxtamedullary nephron and its blood supply.
The tubules and the blood vessels are shown separately for clarity. In the kidney, the peritubular capillary network surrounds the convoluted tubules, and the distal convoluted tubule passes close to the glomerulus, between the afferent and efferent arterioles. (This last is shown in more detail in Fig. 28.2.)
The Blood Supply to the Nephron
Nephrons possess the special characteristic of having two capillary beds in series with each other (see Fig. 28.1). The afferent arteriole of each cortical nephron branches to form the glomerulus; glomerular capillaries coalesce into the efferent arteriole which, in turn, branches to form a second capillary network in the cortex, around the convoluted tubules and loops of Henle, before converging on venules and thence on renal veins. By contrast, efferent arterioles of juxtamedullary nephrons lead to vessel loops (vasa recta) that pass deep into the medulla with the thin loops of Henle, and play a key role in counter-current exchange (see below).
The Juxtaglomerular Apparatus
A conjunction of afferent arteriole, efferent arteriole and distal convoluted tubule near the glomerulus forms the juxtaglomerular apparatus (Fig. 28.2). At this site, there are specialised cells in both the afferent arteriole and in the tubule. The latter, termed macula densa cells, respond to changes in the rate of flow and the composition of tubule fluid, and they control renin release from specialised granular renin-containing cells in the afferent arteriole (Ch. 22). Other mediators also influence renin secretion, including β2 agonists, vasodilator prostaglandins and feedback inhibition from angiotensin II acting on AT1 receptors (see Fig. 22.4). The role of the juxtaglomerular apparatus in the control of Na+ balance is dealt with below.
Fig. 28.2 The juxtaglomerular apparatus.
The cutaway sections show the granular renin-containing cells round the afferent arteriole, and the macula densa cells in the distal convoluted tubule. The inset shows the general relationships between the structures. DT, distal tubule; G, glomerulus.
(Modified from Sullivan & Grantham, 1982.)
Glomerular Filtration
Fluid is driven from the capillaries into the tubular capsule (Bowman’s capsule) by hydrodynamic force opposed by the oncotic pressure of the plasma proteins, to which the glomerular capillaries are impermeable. All the low-molecular-weight constituents of plasma appear in the filtrate, while albumin and larger proteins are retained in the blood.
Tubular Function
The apex (lumenal surface) of each tubular cell is surrounded by a tight junction, as in all epithelia. This is a specialised region of membrane that separates the intercellular space from the lumen. The movement of ions and water across the epithelium can occur through cells (the transcellular pathway) and between cells through the tight junctions (the paracellular pathway). A common theme is that energy is expended to pump Na+ out of the cell by Na+-K+-ATPase situated in the basolateral cell membrane and the resulting gradient of Na+ concentration drives the entry of Na+ from the lumen via various transporters that facilitate Na+ entry coupled with movement of other ions. These transporters vary in different parts of the nephron as described below.
The Proximal Convoluted Tubule
The epithelium of the proximal convoluted tubule is ‘leaky’, i.e. the tight junctions in the proximal tubule are not so ‘tight’ after all, being permeable to ions and water, and permitting passive flow in either direction. This prevents the build-up of large concentration gradients; thus, although approximately 60–70% of Na+ reabsorption occurs in the proximal tubule, this transfer is accompanied by passive absorption of water so that fluid leaving the proximal tubule remains approximately isotonic to the filtrate entering Bowman’s capsule.
Some of the transport processes in the proximal tubule are shown in Figures 28.3-28.5. The most important mechanism for Na+ entry into proximal tubular cells from the filtrate occurs by Na+/H+ exchange (Fig. 28.5). Intracellular carbonic anhydrase is essential for production of H+ for secretion into the lumen. Na+ is reabsorbed in exchange for H+, and transported out of the cells into the interstitium and thence into the blood by a Na+-K+-ATPase (sodium pump) in the basolateral membrane. This is the main active transport mechanism of the nephron in terms of energy consumption.
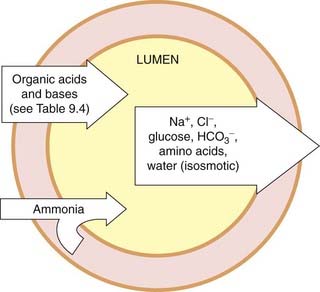
Fig. 28.3 Transport processes in the proximal convoluted tubule.
The main driving force for the absorption of solutes and water from the lumen is the Na+-K+-ATPase in the basolateral membrane of the tubule cells. Many drugs are secreted into the proximal tubule (see Ch. 9).
(Redrawn from Burg, 1985.)
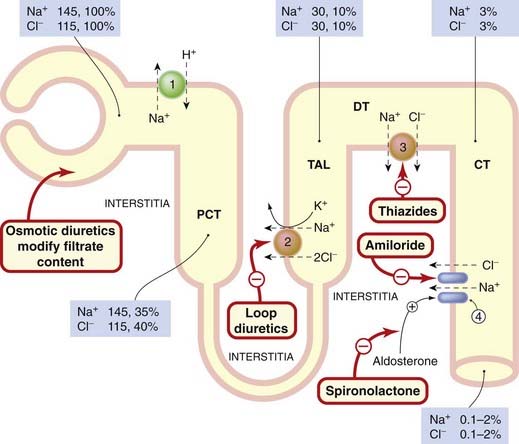
Fig. 28.4 Schematic showing the absorption of sodium and chloride in the nephron and the main sites of action of drugs.
Cells are depicted as an orange border round the yellow tubular lumen. Mechanisms of ion absorption at the apical margin of the tubule cell: (1) Na+/H+ exchange; (2) Na+/K+/2Cl− co-transport; (3) Na+/Cl− co-transport; (4) Na+ entry through sodium channels. Sodium is pumped out of the cells into the interstitium by the Na+-K+-ATPase in the basolateral margin of the tubular cells (not shown). The numbers in the boxes give the concentration of ions as millimoles per litre of filtrate, and the percentage of filtered ions still remaining in the tubular fluid at the sites specified. CT, collecting tubule; DT, distal tubule; PCT, proximal convoluted tubule; TAL, thick ascending loop.
(Data from Greger, 2000.)
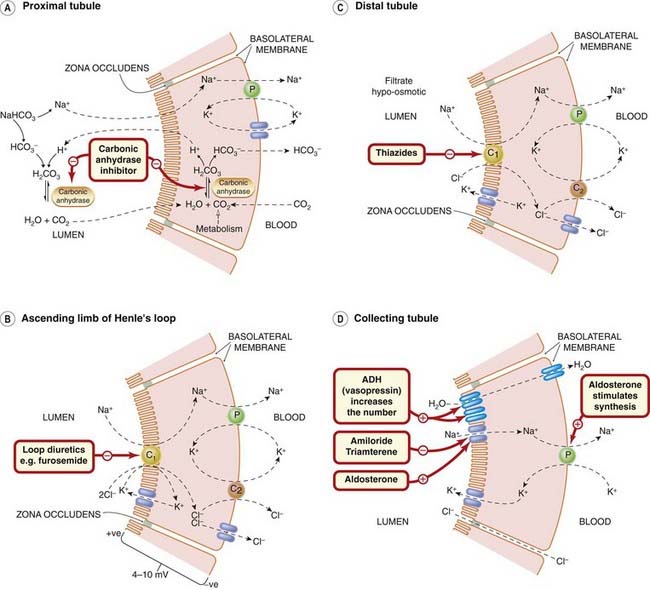
Fig. 28.5 Drug effects on renal tubular ion transport.
The primary active transport mechanism is the Na+ pump (P) in the basolateral membrane. The diagram is simplified: the Na+ pump (in each panel) exchanges 3Na+ for 2K+. [A] Bicarbonate ion reabsorption in the proximal convoluted tubule, showing the action of carbonic anhydrase inhibitors. Sodium ions are absorbed and H+ secreted at the lumenal surface by an antiport mechanism (Na+/H+ exchanger). [B] Ion transport in the thick ascending limb of Henle’s loop, showing the site of action of loop diuretics. Na+, K+ and Cl− enter by a co-transport system (C1). Chloride leaves the cell both through basolateral chloride channels and by an electroneutral K+/Cl− co-transport system (C2). Some K+ returns to the lumen via potassium channels in the apical membrane, and some Na+ is absorbed paracellularly through the zonula occludens. [C] Salt transport in the distal convoluted tubule, showing the site of action of thiazide diuretics. Sodium and chloride ions enter by an electroneutral co-transport carrier (C1). Some Cl− is transported out of the cell by a K+/Cl− co-transport carrier (C2); some leaves the cell through chloride channels. Some K+ is transported out of the cell by the co-transport carrier (C2), and some passes back into the tubule lumen through potassium channels. [D] Actions of hormones and drugs on the collecting tubule. The cells are impermeable to water in the absence of antidiuretic hormone (ADH), and to Na+ in the absence of aldosterone. Aldosterone acts on a nuclear receptor within the tubule cell and on membrane receptors. Chloride ions exit the tubule through the paracellular pathway. Potassium ions are added to the filtrate, as is H+ (not shown).
(Adapted from Greger, 2000.)
 Bicarbonate is normally completely reabsorbed in the proximal tubule. This is achieved by combination with protons, yielding carbonic acid, which dissociates to form carbon dioxide and water—a reaction catalysed by carbonic anhydrase present in the lumenal brush border of the proximal tubule cells (Fig. 28.5A)—followed by passive reabsorption of the dissolved carbon dioxide.1 The selective removal of sodium bicarbonate, with accompanying water, in the early proximal tubule causes a secondary rise in the concentration of chloride ions. Diffusion of chloride down its concentration gradient via the paracellular shunt leads, in turn, to a lumen-positive potential difference that favours reabsorption of sodium. The other mechanism involved in movement via the paracellular route is that sodium ions are secreted by Na+-K+-ATPase into the lateral intercellular space, slightly raising its osmolality because of its 3 : 2 stoichiometry. This leads to osmotic movement of water across the tight junction, in turn causing sodium reabsorption by convection (solvent drag).
Bicarbonate is normally completely reabsorbed in the proximal tubule. This is achieved by combination with protons, yielding carbonic acid, which dissociates to form carbon dioxide and water—a reaction catalysed by carbonic anhydrase present in the lumenal brush border of the proximal tubule cells (Fig. 28.5A)—followed by passive reabsorption of the dissolved carbon dioxide.1 The selective removal of sodium bicarbonate, with accompanying water, in the early proximal tubule causes a secondary rise in the concentration of chloride ions. Diffusion of chloride down its concentration gradient via the paracellular shunt leads, in turn, to a lumen-positive potential difference that favours reabsorption of sodium. The other mechanism involved in movement via the paracellular route is that sodium ions are secreted by Na+-K+-ATPase into the lateral intercellular space, slightly raising its osmolality because of its 3 : 2 stoichiometry. This leads to osmotic movement of water across the tight junction, in turn causing sodium reabsorption by convection (solvent drag).
Many organic acids and bases are actively secreted into the tubule from the blood by specific transporters (see below, Fig. 28.3 and Ch. 9).
After passage through the proximal tubule, tubular fluid (now 30–40% of the original volume of the filtrate) passes on to the loop of Henle.
The Loop of Henle, Medullary Counter-Current Multiplier and Exchanger
The loop of Henle consists of a descending and an ascending portion (Figs 28.1 and 28.4), the ascending portion having both thick and thin segments. This part of the nephron enables the kidney to excrete urine that is either more or less concentrated than plasma, and hence to regulate the osmotic balance of the body as a whole. The loops of Henle of the juxtamedullary nephrons function as counter-current multipliers, and the vasa recta as counter-current exchangers. NaCl is actively reabsorbed in the thick ascending limb, causing hypertonicity of the interstitium. In the descending limb, water moves out and the tubular fluid becomes progressively more concentrated as it approaches the bend.
The descending limb is permeable to water, which exits passively because the interstitial fluid of the medulla is kept hypertonic by the counter-current concentrating system. In juxtamedullary nephrons with long loops, there is extensive movement of water out of the tubule so that the fluid eventually reaching the tip of the loop has a high osmolality—normally approximately 1200 mosmol/kg, but up to 1500 mosmol/kg under conditions of dehydration—compared with plasma and extracellular fluid, which is approximately 300 mosmol/kg.2 The hypertonic milieu of medulla, through which the collecting ducts of all nephrons pass on the way to the renal pelvis, is important in providing a mechanism by which the osmolarity of the urine is controlled.
The ascending limb has very low permeability to water, i.e. the tight junctions really are ‘tight’, enabling the build-up of a substantial concentration gradient across the wall of the tubule. It is here, in the thick ascending limb of the loop of Henle, that 20–30% of filtered Na+ is reabsorbed. There is active reabsorption of NaCl, unaccompanied by water, reducing the osmolarity of the tubular fluid and making the interstitial fluid of the medulla hypertonic. The osmotic gradient in the medullary interstitium is the key consequence of the counter-current multiplier system, the main principle being that small horizontal osmotic gradients ‘stack up’ to produce a large vertical gradient. Urea contributes to the gradient because it is more slowly reabsorbed than water and may be added to fluid in the descending limb, so its concentration rises along the nephron until it reaches the collecting tubules, where it diffuses out into the interstitium. It is thus ‘trapped’ in the inner medulla.
Ions move into cells of the thick ascending limb of the loop of Henle across the apical membrane by a Na+/K+/2Cl− co-transporter, driven by the Na+ gradient produced by Na+-K+-ATPase in the basolateral membrane (Fig. 28.5B). Most of the K+ taken into the cell by the Na+/K+/2Cl− co-transporter returns to the lumen through apical potassium channels, but some K+ is reabsorbed, along with Mg2+ and Ca2+.
Reabsorption of salt from the thick ascending limb is not balanced by reabsorption of water, so tubular fluid is hypotonic with respect to plasma as it enters the distal convoluted tubule (Fig. 28.4). The thick ascending limb is therefore sometimes referred to as the ‘diluting segment’.
The Distal Tubule
In the early distal tubule, NaCl reabsorption, coupled with impermeability of the zonula occludens to water, further dilutes the tubular fluid. Transport is driven by Na+-K+-ATPase in the basolateral membrane. This lowers cytoplasmic Na+ concentration, and consequently Na+ enters the cell from the lumen down its concentration gradient, accompanied by Cl−, by means of a Na+/Cl− co-transporter (Fig. 28.5C).
The excretion of Ca2+ is regulated in this part of the nephron, parathormone and calcitriol both increasing Ca2+ reabsorption (see Ch. 35).
The Collecting Tubule and Collecting Duct
Distal convoluted tubules empty into collecting tubules, which coalesce to form collecting ducts (Fig. 28.1). Collecting tubules include principal cells, which reabsorb Na+ and secrete K+ (Fig. 28.5D), and two populations of intercalated cells, α and β, which secrete acid and base, respectively.
The tight junctions in this portion of the nephron are impermeable to water and ions. The movement of ions and water in this segment is under independent hormonal control: absorption of NaCl by aldosterone (Ch. 22), and absorption of water by antidiuretic hormone (ADH), also termed vasopressin (Ch. 32).
Aldosterone enhances Na+ reabsorption and promotes K+ excretion. It promotes Na+ reabsorption by:
ADH and nephrogenic diabetes insipidus
ADH is secreted by the posterior pituitary (Ch. 32) and binds V2 receptors in the basolateral membranes of cells in the collecting tubules and ducts, increasing expression of aquaporin (water channels; see Ch. 8) in the apical membranes (Fig. 28.5D). This renders this part of the nephron permeable to water, allowing passive reabsorption of water as the collecting duct traverses the hyperosmotic region of the medulla, and hence the excretion of concentrated urine. Conversely, in the absence of ADH, collecting duct epithelium is impermeable to water, so hypotonic fluid that leaves the distal tubule remains hypotonic as it passes down the collecting ducts, leading to the excretion of dilute urine. Defective ADH secretion (Ch. 32) or action on the kidney results in diabetes insipidus, a disorder in which patients excrete large volumes of dilute urine.
Ethanol (Ch. 48) inhibits the secretion of ADH, causing a water diuresis (possibly familiar to some of our readers) as a kind of transient diabetes insipidus. Nicotine enhances ADH secretion (perhaps contributing to the appeal of an after-dinner cigar?).
Several drugs inhibit the action of ADH: lithium (used in psychiatric disorders; see Ch. 45), demeclocycline (a tetracycline used not as an antibiotic, but rather to treat inappropriate secretion of ADH from tumours or in other conditions), colchicine (Ch. 26) and vinca alkaloids (Ch. 55). Recently, more specific antagonists of ADH (e.g. conivaptan, tolvaptan) have been introduced for treatment of hyponatraemia (see Ch. 22). All these drugs can cause acquired forms of nephrogenic diabetes insipidus, caused by a failure of the renal collecting ducts to respond to ADH. Nephrogenic diabetes insipidus can also be caused by two genetic disorders affecting the V1 receptor or aquaporin.
Renal tubular function 
Acid–Base Balance
The kidneys (together with the lungs; Ch. 27) regulate the H+ concentration of body fluids. Acid or alkaline urine can be excreted according to need, the usual requirement being to form acid urine to eliminate phosphoric and sulfuric acids generated during the metabolism of nucleic acids, and sulfur-containing amino acids consumed in the diet. Consequently, metabolic acidosis is a common accompaniment of renal failure. Altering urine pH to alter drug exretion is mentioned below.
Potassium Balance
Extracellular K+ concentration—critically important for excitable tissue function (see Ch. 4)—is tightly controlled through regulation of K+ excretion by the kidney. Urinary K+ excretion matches dietary intake, usually approximately 50–100 mmol in 24 h in Western countries. Most diuretics cause K+ loss (see below). This can cause problems if they are co-administered with cardiac glycosides or class III antidysrhythmic drugs whose toxicity is increased by low plasma K+ (Ch. 22)—clinically important drug interactions (see Ch. 56).
Potassium ions are transported into collecting duct and collecting tubule cells from blood and interstitial fluid by Na+-K+-ATPase in the basolateral membrane, and leak into the lumen through a K+-selective ion channel. Na+ passes from tubular fluid through sodium channels in the apical membrane down the electrochemical gradient created by the Na+-K+-ATPase; a lumen-negative potential difference across the cell results, increasing the driving force for K+ secretion into the lumen. Thus K+ secretion is coupled to Na+ reabsorption.
Excretion of Organic Molecules
There are distinct mechanisms (see Ch. 9, Table 9.4) for secreting organic anions and cations into the proximal tubular lumen. Secreted anions include several important drugs, for example thiazides, furosemide, salicylate (Ch. 26), and most penicillins and cephalosporins (Ch. 50). Similarly, several secreted organic cations are important drugs, for example triamterene, amiloride, atropine (Ch. 13), morphine (Ch. 41) and quinine (Ch. 53). Both anion and cation transport mechanisms are, like other renal ion transport processes, indirectly powered by active transport of Na+ and K+, the energy being derived from Na+-K+-ATPase in the basolateral membrane.
Organic anions are exchanged with α-ketoglutarate by an antiport in the basolateral membrane, and diffuse passively into the tubular lumen (Fig. 28.3).
Organic cations diffuse into the cell from the interstitium and are then actively transported into the tubular lumen in exchange for H+.
Natriuretic Peptides
Endogenous A, B and C natriuretic peptides (ANP, BNP and CNP; see Chs 21 and 22) are involved in the regulation of Na+ excretion. They are released from the heart in response to stretch (A and B), from endothelium (C) and from brain (B). They activate guanylyl cyclase (Ch. 3), and cause natriuresis both by renal haemodynamic effects (increasing glomerular capillary pressure by dilating afferent and constricting efferent arterioles) and by direct tubular actions. The tubular actions include the inhibition of angiotensin II-stimulated Na+ and water reabsorption in the proximal convoluted tubule, and of the action of ADH in promoting water reabsorption in the collecting tubule.
Within the kidney, the post-translational processing of ANP prohormone differs from that in other tissues, resulting in an additional four amino acids being added to the amino terminus of ANP to yield a related peptide, urodilatin, that promotes Na+ excretion by acting on receptors on the lumenal side of the collecting duct cells.
Prostaglandins and Renal Function
Prostaglandins (PGs; see Ch. 17) generated in the kidney modulate its haemodynamic and excretory functions. The main renal prostaglandins in humans are vasodilator and natriuretic, namely PGE2 in the medulla and PGI2 (prostacyclin) in glomeruli. Factors that stimulate their synthesis include ischaemia, angiotensin II, ADH and bradykinin.
Prostaglandin biosynthesis is low under basal conditions. However, when vasoconstrictors (e.g. angiotensin II, noradrenaline) are released, PGE2 and PGI2 modulate their effects on the kidney by causing compensatory vasodilatation.
The influence of renal prostaglandins on salt balance and haemodynamics can be inferred from the effects of non-steroidal anti-inflammatory drugs (NSAIDs, which inhibit prostaglandin production; see Ch. 26). NSAIDs have little or no effect on renal function in healthy people, but predictably cause acute renal failure in clinical conditions in which renal blood flow depends on vasodilator prostaglandin biosynthesis. These include cirrhosis of the liver, heart failure, nephrotic syndrome, glomerulonephritis and extracellular volume contraction (see Ch. 57, Table 57.1). NSAIDs increase blood pressure in patients treated for hypertension by impairing PG-mediated vasodilatation and salt excretion. They exacerbate salt and water retention in patients with heart failure (see Ch. 22), partly by this same direct mechanism.4
Drugs Acting on the Kidney
Diuretics
Diuretics increase the excretion of Na+ and water. They decrease the reabsorption of Na+ and (usually) Cl− from the filtrate, increased water loss being secondary to the increased excretion of NaCl (natriuresis). This can be achieved:
Because a very large proportion of salt (NaCl) and water that passes into the tubule via the glomerulus is reabsorbed (Table 28.1), a small decrease in reabsorption can cause a marked increase in Na+ excretion. A summary diagram of the mechanisms and sites of action of various diuretics is given in Figure 28.4 and more detailed information on different classes of drugs in Figure 28.5.
Most diuretics with a direct action on the nephron act from within the tubular lumen and reach their sites of action by being secreted into the proximal tubule (spironolactone is an exception).
Diuretics Acting Directly on Cells of the Nephron
The main therapeutically useful diuretics act on the:
For a more detailed review of the actions and clinical uses of the diuretics, see Greger et al. (2005).
Loop diuretics
Loop diuretics (Fig. 28.5B) are the most powerful diuretics (see Fig. 28.6 for a comparison with thiazides), capable of causing the excretion of 15–25% of filtered Na+. Their action is often described—in a phrase that conjures up a rather uncomfortable picture—as causing ‘torrential urine flow’. The main example is furosemide; bumetanide is an alternative agent. These drugs act on the thick ascending limb, inhibiting the Na+/K+/2Cl− carrier in the lumenal membrane by combining with its Cl− binding site.
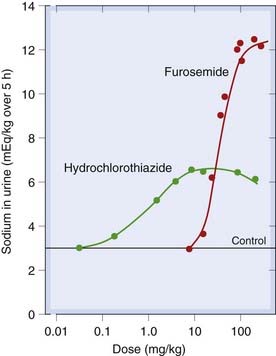
Fig. 28.6 Dose–response curves for furosemide (frusemide) and hydrochlorothiazide, showing differences in potency and maximum effect ‘ceiling’.
Note that these doses are not used clinically.
(Adapted from Timmerman R J et al. 1964 Curr Ther Res 6: 88.)
Loop diuretics also have incompletely understood vascular actions. Intravenous administration of furosemide to patients with pulmonary oedema caused by acute heart failure (see Ch. 22) causes a therapeutically useful vasodilator effect before the onset of diuresis. Possible mechanisms that have been invoked include decreased vascular responsiveness to vasoconstrictors such as angiotensin II and noradrenaline; increased formation of vasodilating prostaglandins (see above); decreased production of the endogenous ouabain-like natriuretic hormone (Na+-K+-ATPase inhibitor; see Ch. 21), which has vasoconstrictor properties; and potassium channel-opening effects in resistance arteries (see Greger et al., 2005).
Loop diuretics increase the delivery of Na+ to the distal nephron, causing loss of H+ and K+. Because Cl− but not HCO3− is lost in the urine, the plasma concentration of HCO3− increases as plasma volume is reduced—a form of metabolic alkalosis therefore referred to as ‘contraction alkalosis’.
Loop diuretics increase excretion of Ca2+ and Mg2+ and decrease excretion of uric acid.
Pharmacokinetic aspects
Loop diuretics are absorbed from the gastrointestinal tract, and are usually given by mouth. They may also be given intravenously in urgent situations (e.g. acute pulmonary oedema) or when intestinal absorption is impaired, for example as a result of reduced intestinal perfusion in patients with chronic congestive heart failure, who can become resistant to the action of orally administered diuretics. Given orally, they act within 1 h; given intravenously, they produce a peak effect within 30 min. Loop diuretics are strongly bound to plasma protein, and so do not pass directly into the glomerular filtrate. They reach their site of action—the lumenal membrane of the cells of the thick ascending limb—by being secreted in the proximal convoluted tubule by the organic acid transport mechanism; the fraction thus secreted is excreted in the urine.
In nephrotic syndrome,5 loop diuretics become bound to albumin in the tubular fluid, and consequently are not available to act on the Na+/K+/2Cl− carrier—another cause of diuretic resistance. Molecular variation in the Na+/K+/2Cl− carrier may also be important in some cases of diuretic resistance (Shankar & Brater, 2003).
The fraction not excreted in the urine is metabolised, mainly in liver—bumetanide by cytochrome P450 pathways and furosemide being glucuronidated. The plasma half-lives of both these drugs are approximately 90 min (longer in renal failure), and the durations of action 3–6 h. The clinical use of loop diuretics is given in the box.
Clinical uses of loop diuretics (e.g. furosemide) 
Unwanted effects
Unwanted effects directly related to the renal action of loop diuretics are common.6 Excessive Na+ and water loss are common, especially in elderly patients, and can cause hypovolaemia and hypotension. Potassium loss, resulting in low plasma K+ (hypokalaemia), and metabolic alkalosis are common. Hypokalaemia increases the effects and toxicity of several drugs (e.g. digoxin and type III antidysrhythmic drugs, Ch. 21), so this is potentially a clinically important source of drug interaction (Ch. 56). If necessary, hypokalaemia can be averted or treated by concomitant use of K+-sparing diuretics (see below), sometimes with supplementary potassium replacement. Hypomagnesaemia is less often recognised but can also be clinically important. Hyperuricaemia is common and can precipitate acute gout (see Ch. 26). Excessive diuresis leads to reduced renal perfusion and pre-renal renal impairment (an early warning of this is a rise in serum urea concentration).
Unwanted effects unrelated to the renal actions of the drugs are infrequent. Dose-related hearing loss (compounded by concomitant use of other ototoxic drugs such as aminoglycoside antibiotics) can result from impaired ion transport by the basolateral membrane of the stria vascularis in the inner ear. It occurs only at much higher doses than usually needed to produce diuresis. Idiosyncratic allergic reactions (e.g. rashes, bone marrow depression) are uncommon.
Diuretics acting on the distal tubule
Diuretics acting on the distal tubule include thiazides (e.g. bendroflumethiazide, hydrochlorothiazide) and related drugs (e.g. chlortalidone, indapamide and metolazone; see Fig. 28.5C).
Thiazides are less powerful than loop diuretics but are preferred in treating uncomplicated hypertension (Ch. 22). They are better tolerated than loop diuretics, and in clinical trials have been shown to reduce risks of stroke and heart attack associated with hypertension. In the largest trial (ALLHAT 2002), chlortalidone performed as well as newer antihypertensive drugs (an angiotensin-converting enzyme [ACE] inhibitor and a calcium antagonist). They bind to the Cl− site of the distal tubular Na+/Cl− co-transport system, inhibiting its action and causing natriuresis with loss of sodium and chloride ions in the urine. The resulting contraction in blood volume stimulates renin secretion, leading to angiotensin formation and aldosterone secretion (Ch. 22, see Figs 22.4 and 22.9). This homeostatic mechanism limits the effect of the diuretic on the blood pressure, resulting in an in vivo dose–hypotensive response relationship with only a gentle gradient during chronic dosing.
Effects of thiazides on Na+, K+, H+ and Mg2+ balance are qualitatively similar to those of loop diuretics, but smaller in magnitude. In contrast to loop diuretics, however, thiazides reduce Ca2+ excretion, which may be advantageous in older patients at risk of osteoporosis. This could favour thiazides over loop diuretics in terms of bone metabolism during long-term use in older patients (Schoofs et al., 2003).
Although thiazides are milder than loop diuretics when used alone, co-administration with loop diuretics has a synergistic effect, because the loop diuretic delivers a greater fraction of the filtered load of Na+ to the site of action of the thiazide in the distal tubule.
Thiazide diuretics have a vasodilator action (see Chs 4 and 22). When used in the treatment of hypertension (Ch. 22), the initial fall in blood pressure results from the decreased blood volume caused by diuresis, but vasodilatation contributes to the later phase.
Thiazide diuretics have a paradoxical effect in diabetes insipidus, where they reduce the volume of urine by interfering with the production of hypotonic fluid in the distal tubule, and hence reduce the ability of the kidney to excrete hypotonic urine (i.e. they reduce free water clearance).
Pharmacokinetic aspects
Thiazides and related drugs are effective orally. All are excreted in the urine, mainly by tubular secretion, and they compete with uric acid for the organic anion transporter (OAT; see Ch. 9). Bendroflumethiazide has its maximum effect at about 4–6 h and duration is 8–12 h. Chlortalidone has a longer duration of action.
The clinical use of thiazide diuretics is given in the clinical box.
Unwanted effects
Apart from an increase in urinary frequency, the commonest unwanted effect (not obviously related to the main renal actions of the thiazides) is erectile dysfunction. This emerged in an analysis of reasons given by patients for withdrawing from blinded treatment in the Medical Research Council mild hypertension trial, where (to the surprise of the investigators) erectile dysfunction was substantially more common than in men allocated to a β-adrenoceptor antagonist or to placebo. Thiazide-associated erectile dysfunction is reversible; it is less common with the low doses used in current practice but remains a problem. Potassium loss can be important, as can loss of Mg2+. Excretion of uric acid is decreased, and hypochloraemic alkalosis can occur.
Impaired glucose tolerance (see Ch. 30), due to inhibition of insulin secretion, is thought to result from activation of KATP channels in pancreatic islet cells.7 Diazoxide, a non-diuretic thiazide, also activates KATP channels, causing vasodilatation and impaired insulin secretion. Indapamide is said to lower blood pressure with less metabolic disturbance than related drugs, possibly because it is marketed at a lower equivalent dose.
Hyponatraemia is potentially serious, especially in the elderly. Hypokalaemia can be a cause of adverse drug interaction (see above under Loop diuretics) and can precipitate encephalopathy in patients with severe liver disease.
Idiosyncratic reactions (e.g. rashes, blood dyscrasias) are rare but can be serious.
Aldosterone antagonists
Spironolactone and eplerenone (Weinberger, 2004) have very limited diuretic action when used singly, because distal Na+/K+ exchange—the site on which they act (Fig. 28.5D)—accounts for reabsorption of only 2% of filtered Na+. They do, however, have marked antihypertensive effects (Ch. 22), prolong survival in selected patients with heart failure (Ch. 22) and can prevent hypokalaemia when combined with loop diuretics or with thiazides. They compete with aldosterone for its intracellular receptor (see Ch. 32), thereby inhibiting distal Na+ retention and K+ secretion (see Fig. 28.5D).
Pharmacokinetic aspects
Spironolactone is well absorbed from the gut. Its plasma half-life is only 10 min, but its active metabolite, canrenone, has a plasma half-life of 16 h. The action of spironolactone is largely attributable to canrenone. Consistent with this, its onset of action is slow, taking several days to develop. Eplerenone has a shorter elimination half-life than canrenone and has no active metabolites. It is administered by mouth once daily.
Unwanted effects
Aldosterone antagonists predispose to hyperkalaemia, which is potentially fatal. Potassium supplements should not be co-prescribed, and close monitoring of plasma creatinine and electrolytes is needed if these drugs are used for patients with impaired renal function, especially if other drugs that can increase plasma potassium, such as ACE inhibitors, angiotensin receptor antagonists (sartans) (Ch. 22) or β-adrenoceptor antagonists (Ch. 14) are also prescribed—as they often are for patients with heart failure. Gastrointestinal upset is quite common. Actions of spironolactone/canrenone on progesterone and androgen receptors in tissues other than the kidney can result in gynaecomastia, menstrual disorders and testicular atrophy. Eplerenone has lower affinity for these receptors, and such oestrogen-like side effects are less common with licensed doses of this drug.
The clinical use of potassium-sparing diuretics is given in the clinical box.
Clinical uses of potassium-sparing diuretics (e.g. amiloride, spironolactone)
Triamterene and amiloride
Like aldosterone antagonists, triamterene and amiloride have only limited diuretic efficacy, because they also act in the distal nephron, where only a small fraction of Na+ reabsorption occurs. They act on the collecting tubules and collecting ducts, inhibiting Na+ reabsorption by blocking lumenal sodium channels (see Ch. 4) and decreasing K+ excretion (see Fig. 28.5D).
They can be given with loop diuretics or thiazides in order to maintain potassium balance.
Pharmacokinetic aspects
Triamterene is well absorbed in the gastrointestinal tract. Its onset of action is within 2 h, and its duration of action 12–16 h. It is partly metabolised in the liver and partly excreted unchanged in the urine. Amiloride is less well absorbed and has a slower onset, with a peak action at 6 h and duration of about 24 h. Most of the drug is excreted unchanged in the urine.
Unwanted effects
The main unwanted effect, hyperkalaemia, is related to the pharmacological action of these drugs and can be dangerous, especially in patients with renal impairment or receiving other drugs that can increase plasma K+ (see above). Gastrointestinal disturbances have been reported but are infrequent. Triamterene has been identified in kidney stones, but its aetiological role is uncertain. Idiosyncratic reactions, for example rashes, are uncommon.
Carbonic anhydrase inhibitors
Carbonic anhydrase inhibitors (Fig. 28.5A)—for example acetazolamide—increase excretion of bicarbonate with accompanying Na+, K+ and water, resulting in an increased flow of an alkaline urine and metabolic acidosis. These agents, although not now used as diuretics, are still used in the treatment of glaucoma to reduce the formation of aqueous humour (Ch. 13), and also in some types of infantile epilepsy (Ch. 44).
Urinary loss of bicarbonate depletes extracellular bicarbonate, and the diuretic effect of carbonic anhydrase inhibitors is consequently self-limiting.
Diuretics That Act Indirectly by Modifying the Content of the Filtrate
Osmotic diuretics
Osmotic diuretics are pharmacologically inert substances (e.g. mannitol) that are filtered in the glomerulus but not reabsorbed by the nephron (see Fig. 28.4).8 To cause a diuresis, they must constitute an appreciable fraction of the osmolarity of tubular fluid. Within the nephron, their main effect is exerted on those parts of the nephron that are freely permeable to water: the proximal tubule, descending limb of the loop and (in the presence of ADH; see above) the collecting tubules. Passive water reabsorption is reduced by the presence of non-reabsorbable solute within the tubule; consequently a larger volume of fluid remains within the proximal tubule. This has the secondary effect of reducing Na+ reabsorption.
Therefore the main effect of osmotic diuretics is to increase the amount of water excreted, with a smaller increase in Na+ excretion. They are sometimes used in acute renal failure, which can occur as a result of haemorrhage, injury or systemic infections. Glomerular filtration rate is reduced, and absorption of NaCl and water in the proximal tubule becomes almost complete, so that more distal parts of the nephron virtually ‘dry up’, and urine flow ceases. Protein is deposited in the tubules and may impede the flow of fluid. Osmotic diuretics (e.g. mannitol given intravenously in a dose of 12–15 g) can limit these effects, at least if given in the earliest stages, albeit while increasing intravascular volume and risking left ventricular failure.
They are also used for the emergency treatment of acutely raised intracranial or intraocular pressure. Such treatment has nothing to do with the kidney, but relies on the increase in plasma osmolarity by solutes that do not enter the brain or eye, which results in efflux of water from these compartments.
Unwanted effects include transient expansion of the extracellular fluid volume (with a risk of causing left ventricular failure) and hyponatraemia. Headache, nausea and vomiting can occur.
Diuretics
Drugs That Alter the pH of the Urine
It is possible, by the use of pharmacological agents, to produce urinary pH values ranging from approximately 5 to 8.5.
Carbonic anhydrase inhibitors increase urinary pH by blocking bicarbonate reabsorption (see above). Citrate (given by mouth as a mixture of sodium and potassium salts) is metabolised via the Krebs cycle with generation of bicarbonate, which is excreted, alkalinising the urine. This may have some antibacterial effects, as well as improving dysuria (a common symptom of bladder infection, consisting of a burning sensation while passing urine). Additionally, some citrate is excreted in the urine as such and inhibits urinary stone formation. Alkalinisation is important in preventing certain weak acid drugs with limited aqueous solubility, such as sulfonamides (see Ch. 50), from crystallising in the urine; it also decreases the formation of uric acid and cystine stones by favouring the charged anionic form that is more water soluble (Ch. 8).
Alkalinising the urine increases the excretion of drugs that are weak acids (e.g. salicylates and some barbiturates). Sodium bicarbonate is sometimes used to treat salicylate overdose (Ch. 9).
Urinary pH can be decreased with ammonium chloride, but this is now rarely, if ever, used clinically except in a specialised test to discriminate between different kinds of renal tubular acidosis.
Drugs That Alter the Excretion of Organic Molecules
Uric acid metabolism and excretion are relevant in the treatment and prevention of gout (Ch. 26), and a few points about its excretion are made here.
Uric acid is derived from the catabolism of purines, and is present in plasma mainly as ionised urate. In humans, it passes freely into the glomerular filtrate, and most is then reabsorbed in the proximal tubule while a small amount is secreted into the tubule by the anion-secreting mechanism. The net result is excretion of approximately 8–12% of filtered urate. The secretory mechanism is generally inhibited by low doses of drugs that affect uric acid transport (see below), whereas higher doses are needed to block reabsorption. Such drugs therefore tend to cause retention of uric acid at low doses, while promoting its excretion at higher doses. Normal plasma urate concentration is approximately 0.24 mmol/l. In some individuals, the plasma concentration is high, predisposing to gout. In this disorder, urate crystals are deposited in joints and soft tissues,9 resulting in acute arthritis and, if untreated, chronic chalky deposits—‘tophi’—characteristic of this condition. Drugs that increase the elimination of urate (uricosuric agents, e.g. probenecid and sulfinpyrazone) may be useful in such patients, although these have largely been supplanted by allopurinol, which inhibits urate synthesis (Ch. 26).
Probenecid inhibits the reabsorption of urate in the proximal tubule, increasing its excretion. It has the opposite effect on penicillin, inhibiting its secretion into the tubules and raising its plasma concentration. Given orally, probenecid is well absorbed in the gastrointestinal tract, maximal concentrations in the plasma occurring in about 3 h. Approximately 90% is bound to plasma albumin. Free drug passes into the glomerular filtrate but more is actively secreted into the proximal tubule, whence it may diffuse back because of its high lipid solubility (see also Ch. 9). Sulfinpyrazone acts similarly.
The main effect of uricosuric drugs is to block urate reabsorption and lower plasma urate concentration. Both probenecid and sulfinpyrazone inhibit the secretion as well as the reabsorption of urate and, if given in subtherapeutic doses, can actually increase plasma urate concentrations.
Aspirin (and other salicylates such as sulfasalazine), also inhibits urate secretion in normal analgesic doses, increasing blood urate concentration, which may exacerbate gouty arthritis (see Ch. 26). (But salicylates become uricosuric themselves at the very high doses used in the past to treat rheumatoid arthritis.)
Probenicid, as specified above, inhibits penicillin excretion, and at one time was used to enhance the action of penicillin (e.g. in single-dose treatment of gonorrhoea). It is licensed in the UK to prevent nephrotoxicity caused by cidofovir (Ch. 51), an antiviral drug used to treat cytomegalovirus retinitis in AIDS patients for whom other antiviral drugs are inappropriate. It is given with probenecid, and intravenous hydration, to prevent its concentration within the tubular lumen, without which it causes tubular toxicity.
Drugs Used in Renal Failure
Many congenital and acquired diseases damage the kidneys, leading to common end points of acute or chronic renal failure, which are treated by various forms of artificial dialysis or filtration, or renal transplantation. Where possible, treatment of the underlying cause is indicated. Hypertension is both a cause and a consequence of renal impairment, so its treatment with antihypertensive drugs (Ch. 22) is extremely important in the context of renal disease. ACE inhibitors and angiotensin II antagonists have a renoprotective effect—over and above their antihypertensive effect—in some situations. Aggressive management of dyslipidaemia (Ch. 23) is also of great importance. Epoetin (Ch. 24) is used to treat the anaemia of chronic renal failure. Vitamin D preparations (calcitriol or alphacalcidol), used to treat the osteodystrophy of chronic renal failure, are covered in Chapter 35. Antibacterial drugs are crucial in treating renal and urinary tract infections, and are dealt with in Chapter 50.
Renal failure often results in hyperphosphataemia and hyperkalaemia, which may require drug treatment.
Hyperphosphataemia
Phosphate metabolism is closely linked with that of calcium and is discussed in Chapter 35. Phosphate, at concentrations commonly occurring in chronic renal insufficiency, causes vascular smooth muscle cell differentiation into osteoblast-like cells able to sustain mineralisation.
Hyperphosphataemia (plasma phosphate concentration > 1.45 mmol/l) is common in renal failure and may lead calcium phosphate to precipitate in tissues. Large calcium phosphate deposits around joints limit mobility but otherwise cause surprisingly few symptoms. Conjunctival calcification can cause conjunctivitis (‘uraemic red eye’). Calcification of the aortic valve can cause stenosis. Abrupt metastatic calcification in subcutaneous tissues and small vessels can result in extensive soft tissue necrosis (acute calciphylaxis). Hyperphosphataemia is the major trigger for the onset of hyperparathyroidism in early chronic renal failure, and leads to renal osteodystrophy.
These effects of hyperphosphataemia have led to the widespread use of phosphate-binding preparations in renal failure. The antacid aluminium hydroxide (Ch. 29) binds phosphate in the gastrointestinal tract, reducing its absorption, but may increase plasma aluminium in dialysis patients.10 Calcium-based phosphate-binding agents (e.g. calcium carbonate) are widely used. They are contraindicated in hypercalcaemia or hypercalciuria but until recently have been believed to be otherwise safe. However, calcium salts may predispose to tissue calcification (including of artery walls), and calcium-containing phosphate binders may actually contribute to the very high death rates from cardiovascular disease in dialysis patients (Goldsmith et al., 2004).
An anion exchange resin, sevelamer, lowers plasma phosphate, and is less likely than calcium carbonate to cause arterial calcification (Asmus et al., 2005). Sevelamer is not absorbed and has an additional effect in lowering low-density-lipoprotein cholesterol. It is given in gram doses by mouth three times a day with meals. Its adverse effects are gastrointestinal disturbance, and it is contraindicated in bowel obstruction.
Hyperkalaemia
Severe hyperkalaemia is life-threatening. It is commonly caused by renal failure, especially if there is concomitant hypoaldosteronism (e.g. in Addison’s disease; Ch. 32) and by potassium-sparing diuretics (see above) or drugs that interfere with renin secretion (e.g. β-adrenoceptor antagonists; Ch. 14), or with angiotensin II formation or action (i.e. ACE inhibitors and angiotensin receptor antagonists; Ch. 22).
Prompt treatment is indicated if the plasma K+ concentration exceeds 6.5 mmol/l. Cardiac toxicity is counteracted directly by administering calcium gluconate intravenously (Table 21.1), and by measures that shift K+ into the intracellular compartment, for example glucose plus insulin (Ch. 30, clinical box). Salbutamol (albuterol), administered intravenously or by inhalation, also causes cellular K+ uptake and is used for this indication (e.g. Murdoch et al., 1991); it acts synergistically with insulin. Intravenous sodium bicarbonate is also often recommended, and moves potassium into cells. Removal of excessive potassium from the body can be achieved by cation exchange resins such as sodium or calcium polystyrene sulfonate administered by mouth (in combination with sorbitol to prevent constipation) or as an enema. Dialysis is often needed.
Drugs Used in Urinary Tract Disorders
Bed wetting (enuresis) is normal in very young children and persists in around 5% of children aged 10. Disordered micturition is also extremely common in adults of either sex, and becomes more so with advancing age. Associated structural problems (e.g. prostatic hypertrophy, uterine prolapse) may warrant surgical intervention, and urinary infection—curable with antibiotics—may have been overlooked. However, many cases of incontinence (socially devastating) are functional, and should in principle be amenable to drugs acting on urinary tract smooth muscle or on the nerves controlling this. Currently available treatment is, however, disappointing, perhaps because it is not easy to prevent incontinence without causing urinary retention.
Nocturnal enuresis in children aged 10 or more may warrant desmopressin (an analogue of antidiuretic hormone, given by mouth or by nasal spray; Ch. 32) combined with restricting fluid intake, in addition to practical measures such as an enuresis alarm. Tricyclic antidepressants such as amitriptyline (Ch. 46) are sometimes used for up to 3 months, but adverse effects including behaviour disturbance can occur, and relapse is common after stopping treatment.
Symptoms from benign prostatic hypertrophy may be improved by α1-adrenoceptor antagonists, for example doxazosin or tamsulosin (Ch. 14), or by an inhibitor of androgen synthesis such as finasteride (Ch. 34).
Incontinence in adults caused by neurogenic detrusor muscle instability is managed by pelvic floor exercises combined with muscarinic receptor antagonists (Ch. 13) such as oxybutinin, tolterodine, propiverine or trospium, but the dose is limited by their adverse effects.
References and Further Reading
Agre P. Aquaporin water channels (Nobel lecture). Angewandte Chemie—International Edition. 2004;43:4278-4290.
Gamba G. Molecular physiology and pathophysiology of electroneutral cation–chloride cotransporters. Physiol. Rev.. 2005;85:423-493. (Comprehensive review of the molecular biology, structure–function relationships, and physiological and pathophysiological roles of each co-transporter)
Greger R. Physiology of sodium transport Am. J. Med. Sci. 319:2000:51-62 (Outstanding article. Covers not only Na+ transport but also, briefly, that of K+, H+, Cl−, HCO3−, Ca2+, Mg2+ and some organic substances in each of the main parts of the nephron. Discusses regulatory factors, pathophysiological aspects and pharmacological principles)
Lee W., Kim R.B. Transporters and renal drug elimination. Annu. Rev. Pharmacol. Toxicol.. 2003;44:137-166. (Review)
Sullivan L.P., Grantham J.J. The physiology of the kidney, second ed. Philadelphia: Lea & Febiger; 1982.
Greger R., Lang F., Sebekova K., Heidland A. Action and clinical use of diuretics. In: Davison A.M., Cameron J.S., Grunfeld J.P., et al, editors. Oxford textbook of clinical nephrology. third ed. Oxford: Oxford University Press; 2005:2619-2648. (Succinct authoritative account of cellular mechanisms; strong on clinical uses)
Schoofs M.W., van der Klift M., Hofman A., et al. Thiazide diuretics and the risk for hip fracture. Ann. Intern. Med.. 2003;139:476-482. (Rotterdam study: thiazide diuretics protected against hip fracture, but protection disappears after use is discontinued)
Shankar S.S., Brater D.C. Loop diuretics: from the Na–K–2Cl transporter to clinical use Am. J. Physiol. Renal. Physiol. 284:2003:F11-F21 (Reviews pharmacokinetics and pharmacodynamics of loop diuretics in health and in oedematous disorders; the authors hypothesise that altered expression or activity of the Na+/K+/2Cl− transporter possibly accounts for reduced diuretic responsiveness)
Weinberger M.H. Eplerenone—a new selective aldosterone receptor antagonist. Drugs Today. 2004;40:481-485. (Review)
Ca2+/PO4− (see also Diuretics section, above)
Asmus H.G., Braun J., Krause R., et al. Two year comparison of sevelamer and calcium carbonate effects on cardiovascular calcification and bone density. Nephrol. Dial. Transplant.. 2005;20:1653-1661. (Less progression of vascular calcification with sevelamer)
Cozzolino M., Brancaccio D., Gallieni M., Slatopolsky E. Pathogenesis of vascular calcification in chronic kidney disease. Kidney Int.. 2005;68:429-436. (Reviews hyperphosphataemia and hypercalcaemia as independent risk factors for higher incidence of cardiovascular events in patients with chronic kidney disease: ‘… hyperphosphatemia accelerates the progression of secondary hyperparathyroidism with the concomitant bone loss, possibly linked to vascular calcium-phosphate precipitation’)
Goldsmith D., Ritz E., Covic A. Vascular calcification: a stiff challenge for the nephrologist—does preventing bone disease cause arterial disease? Kidney Int.. 2004;66:1315-1333. (Potential danger of using calcium salts as phosphate binders in patients with chronic renal failure)
Antihypertensives and renal protection
ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. JAMA 288:2002:2981-2997 (Massive trial; see also Appel, L.J. for editorial comment: ‘The verdict from ALLHAT—thiazide diuretics are the preferred initial therapy for hypertension’ JAMA 288, 3039–3042)
Nijenhuis T., Vallon V., van der Kemp A.W., et al Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia J. Clin. Invest. 115:2005:1651-1658 (Micropuncture studies in mouse knockouts showing that enhanced passive Ca2+ transport in the proximal tubule rather than active Ca2+ transport in distal convolution explains thiazide-induced hypocalciuria)
Sodium and potassium ion disorders
Coca S.G., Perazella M.A., Buller G.K. The cardiovascular implications of hypokalemia. Am. J. Kidney Dis.. 2005;45:233-247. (The recent discovery that aldosterone antagonists decrease pathological injury of myocardium and endothelium has focused interest on their mechanism; this review addresses the relative benefits of modulating potassium balance versus non-renal effects of aldosterone blockade)
Murdoch I.A., Dos Anjos R., Haycock G.B. Treatment of hyperkalaemia with intravenous salbutamol. Arch. Dis. Child. 1991;66:527-528. (First description of this approach in children)
Drug utilisation in kidney disease
Carmichael D.J.S. Handling of drugs in kidney disease. In: Davison A.M., Cameron J.S., Grunfeld J.P., et al, editors. Oxford textbook of clinical nephrology. third ed. Oxford: Oxford University Press; 2005:2599-2618. (Principles and practice of dose adjustment in patients with renal failure)
1The reaction is reversible, and the enzyme (as any catalyst) does not alter the equilibrium, just speeds up the rate with which it is attained. The concentrations inside the cell are such that carbon dioxide combines with water to produce carbonic acid: the same enzyme (carbonic anhydrase) catalyses this as well (Fig. 28.5A).
2These figures are for humans; some other species, notably the desert rat, can do much better, with urine osmolalities up to 5000 mosmol/kg.
3A mechanism distinct from regulation of gene transcription, which is the normal transduction mechanism for steroid hormones (Ch. 3).
4Additionally, NSAIDs make many of the diuretics used to treat heart failure less effective by competing with them for the organic anion transport (OAT) mechanism mentioned above; loop diuretics and thiazides act from within the lumen by inhibiting exchange mechanisms—see later in this chapter—so blocking their secretion into the lumen reduces their effectiveness by reducing their concentrations at their sites of action.
5Several diseases that damage renal glomeruli impair their ability to retain plasma albumin, causing massive loss of albumin in the urine and a reduced concentration of albumin in the plasma, which can in turn cause peripheral oedema. This is referred to as nephrotic syndrome.
6Such unwanted effects are re-enacted in extreme form in Bartter syndrome type 1, a rare autosomal recessive single gene disorder of the Na+/K+/2Cl− transporter, whose features include polyhydramnios—caused by fetal polyuria—and, postnatally, renal salt loss, low blood pressure, hypokalaemic metabolic alkalosis and hypercalciuria.
7The chemically related sulfonylurea group of drugs used to treat diabetes mellitus (Ch. 30) act in the opposite way, by closing KATP channels and enhancing insulin secretion.
8In hyperglycaemia, glucose acts as an osmotic diuretic once plasma glucose exceeds the renal reabsorptive threshold (usually approximately 12 mmol/l), accounting for the cardinal symptom of polyuria in diabetes mellitus; see Chapter 30.
9The distribution is determined by body temperature: crystals come out of solution in cool extremities such as the joints of the big toe—the classic site for acute gout—and the pinna of the ear, a common site for gouty tophi.
10Before Kerr identified the cause in Newcastle, the use of alum to purify municipal water supplies led to a horrible and untreatable neurodegenerative condition known as ‘dialysis dementia’, and also to a particularly painful and refractory form of bone disease.