DYSREGULATION OF CANCER-ASSOCIATED GENES
The genetic damage that activates oncogenes or inactivates tumor suppressor genes may be subtle (e.g., point mutations) or may involve segments of chromosomes large enough to be detected in a routine karyotype. Activation of oncogenes and loss of function of tumor suppressor genes by mutations were discussed earlier in this chapter. Here we discuss chromosomal abnormalities. We end this section by discussing the epigenetic changes that contribute to carcinogenesis.
Chromosomal Changes
In certain neoplasms, karyotypic abnormalities are nonrandom and common. Specific chromosomal abnormalities have been identified in most leukemias and lymphomas, many sarcomas, and an increasing number of carcinomas. In addition, whole chromosomes may be gained or lost. Although changes in chromosome number (aneuploidy) and structure are generally considered to be late phenomena in cancer progression, it has been suggested that aneuploidy and chromosomal instability may be the initiating events in tumor growth.
The study of chromosomal changes in tumor cells is important on two accounts. First, molecular cloning of genes in the vicinity of chromosomal breakpoints or deletions has been extremely useful in identification of oncogenes (e.g., BCL2, ABL) and tumor suppressor genes (e.g., APC, RB). Second, certain karyotypic abnormalities are specific enough to be of diagnostic value, and in some cases they are predictive of clinical course. The translocations associated with the ABL oncogene in CML and with c-MYC in Burkitt lymphoma have been mentioned earlier, in the context of molecular defects in cancer cells (see Fig. 7-27). Several other karyotype alterations in cancer cells are presented in the discussion of specific tumors in later chapters.
Two types of chromosomal rearrangements can activate proto-oncogenes—translocations and inversions. Chromosomal translocations are much more common (Table 7-9) and are discussed here. Translocations can activate proto-oncogenes in two ways:
TABLE 7-9 Selected Examples of Oncogenes Activated by Translocation
| Malignancy | Translocation | Affected Genes* |
|---|---|---|
| Chronic myeloid leukemia | (9;22)(q34;q11) |
 |
| Acute leukemias (AML and ALL) | (8;21)(q22;q22) | AML 8q22 |
| (15;17)(q22;q21) |
 |
|
| Burkitt lymphoma | (8;14)(q24;q32) |
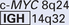 |
| Mantle cell lymphoma | (11;14)(q13;q32) |
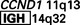 |
| Follicular lymphoma | (14;18)(q32;q21) |
 |
| T-cell ALL | (10;14)(q24;q11) |
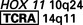 |
| Ewing sarcoma | (11;22)(q24;q12) |
 |
| Prostatic adenocarcinoma | (21;21)(q22;q22) | TMPRSS2 (21q22.3) |
| (7:21)(p22;q22) | ERG (21q22.2) | |
| (17:21)(p21;q22) | ETV1 (7p21.2) | |
| ETV4 (17q21) |
AML, acute myeloid leukemia; ALL, acute lymphoblastic leukemia.
* Genes in boldface are involved in multiple translocations.
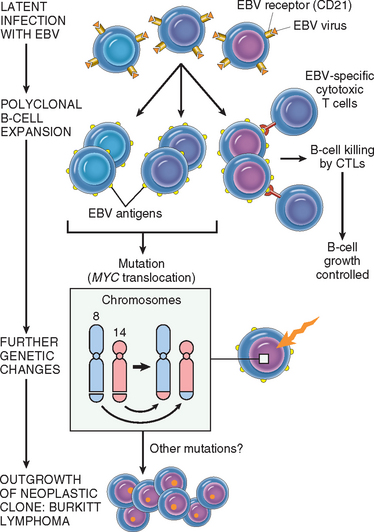
Overexpression of a proto-oncogene caused by translocation is best exemplified by Burkitt lymphoma. All such tumors carry one of three translocations, each involving chromosome 8q24, where the MYC gene has been mapped, as well as one of the three immunoglobulin gene–carrying chromosomes. At its normal locus, MYC is tightly controlled, and is most highly expressed in actively dividing cells. In Burkitt lymphoma the most common form of translocation results in the movement of the MYC-containing segment of chromosome 8 to chromosome 14q32 (see Fig. 7-27), placing it close to the IGH gene. The genetic notation for the translocation is t(8:14)(q24;q32). The molecular mechanisms of the translocation-associated activation of MYC are variable, as are the precise breakpoints within the gene. In most cases the translocation causes mutation or loss of the regulatory sequences of the MYC gene, replacing them with the control regions of the IGH locus, which is highly expressed in B-cell precursors. As the coding sequences remain intact, the gene is constitutively expressed at high levels. The invariable presence of the translocated MYC gene in Burkitt lymphomas attests to the importance of MYC overactivity in the pathogenesis of this tumor.
There are other examples of oncogenes translocated to antigen receptor loci in lymphoid tumors. As mentioned earlier, in mantle cell lymphoma the cyclin D1 gene (CCND1) on chromosome 11q13 is overexpressed by juxtaposition to the IGH locus on 14q32. In follicular lymphomas, a t(14;18)(q32;q21) translocation, the most common translocation in lymphoid malignancies, causes activation of the BCL2 gene. Not unexpectedly, all these tumors in which the immunoglobulin gene is involved are of B-cell origin. In an analogous situation, overexpression of several proto-oncogenes in T-cell tumors results from translocations of oncogenes into the T-cell antigen receptor locus. The affected oncogenes are diverse, but in most cases, as with MYC, they encode nuclear transcription factors.
The Philadelphia chromosome, characteristic of CML and a subset of acute lymphoblastic leukemias, provides the prototypic example of an oncogene formed by fusion of two separate genes. In these cases, a reciprocal translocation between chromosomes 9 and 22 relocates a truncated portion of the proto-oncogene c-ABL (from chromosome 9) to the BCR (breakpoint cluster region) on chromosome 22 (see Fig. 7-27). The hybrid fusion gene BCR-ABL encodes a chimeric protein that has constitutive tyrosine kinase activity. As mentioned, BCR-ABL tyrosine kinase has served as a target for leukemia therapy, with remarkable success so far. Although the translocations are cytogenetically identical in CML and acute lymphoblastic leukemias, they usually differ at the molecular level. In most cases of CML the chimeric protein has a molecular weight of 210 kD, whereas in the more aggressive acute leukemias a 190-kD BCR-ABL fusion protein is typically formed.48,49
Transcription factors are often the partners in gene fusions occurring in cancer cells. For instance, the MLL (myeloid, lymphoid leukemia) gene on 11q23, which itself is a component of a chromatin-remodeling complex, is known to be involved in 50 different translocations with several different partner genes, some of which encode transcription factors (see Table 7-9). Ewing sarcoma/primitive neuroectodermal tumor (PNET) is defined by translocation of the Ewing sarcoma (EWSR1) gene at 22q12, which is involved in numerous translocations, and all of its partner genes analyzed so far also encode a transcription factor. In Ewing sarcoma/PNET, for example, the EWSR1 gene fuses with the FLI1 gene, also a member of the ETS transcription factor family; the resultant chimeric EWS-FLI1 protein has transforming ability. One might ask, why are particular translocations so strongly associated with specific tumors? This is incompletely understood, but one recurrent theme is that at least one of the affected genes often encodes a transcription factor that is required for the development and differentiation of normal cells of the same lineage as the tumor. For example, in acute leukemias many genes involved by recurrent translocations (such as MLL) play essential roles in regulating the self-renewal of hematopoietic stem cells and the normal differentiation of lymphoid and myeloid cells. The fusion proteins resulting from translocations most often inhibit, but occasionally increase, transcriptional function. Until recently, most known translocations were discovered in leukemias/lymphomas and sarcomas; few common translocations had been identified in carcinomas, even though carcinomas are more common. The complex karyotypes of most carcinomas have made identifying translocations difficult. Recently, however, a translocation involving an androgen-regulated gene, TMPRSS2 (21q22), and one of three ETS family transcription factors (ERG [21q22], ETV1 [7p22.2], or ETV4 [17q21]) was found to be present in 50% or more prostate adenocarcinomas.137,138 Development of this translocation seems to occur early in carcinogenesis, in that it is also present in high-grade prostatic intraepithelial neoplasia, a precursor lesion. Although the mechanism by which this translocation causes cancer is not completely understood, it removes the ETS family gene from its normal control region and fuses it to the androgen-regulated TMPRSS2. Thus, the ETS family transcription factor is inappropriately expressed in prostate cells, and as noted above with Ewing sarcoma, when ETS proteins are inappropriately expressed they have transforming ability. There is significant interest in identifying additional fusion genes in other carcinomas. Many fusion genes are thought to be initiators in carcinogenesis, and it is postulated that many cancers may be “addicted” to their properties, similar to the oncogene addiction seen in CML with the BCR-ABL fusion. Thus, inhibition of these genes may provide an avenue for targeted therapy.
Deletions.
Chromosomal deletions are the second most prevalent structural abnormality in tumor cells. Compared with translocations, deletions are more common in nonhematopoietic solid tumors. Deletion of specific regions of chromosomes is associated with the loss of particular tumor suppressor genes. As discussed, deletions involving chromosome 13q14, the site of the RB gene, are associated with retinoblastoma. Deletions of 17p, 5q, and 18q have all been noted in colorectal cancers; these regions harbor three tumor suppressor genes. Deletion of 3p, noted in several tumors, is extremely common in small-cell lung carcinomas, and the hunt is on for one or more cancer suppressor genes at this locale.
Gene Amplification
Activation of proto-oncogenes associated with overexpression of their products may result from reduplication and amplification of their DNA sequences.139 Such amplification may produce several hundred copies of the proto-oncogene in the tumor cell. The amplified genes can be readily detected by molecular hybridization with appropriate DNA probes. In some cases the amplified genes produce chromosomal changes that can be identified microscopically. Two mutually exclusive patterns are seen: multiple small, centric structures called double minutes and homogeneous staining regions. The latter derive from the insertion of the amplified genes into new chromosomal locations, which may be distant from the normal location of the involved genes; because regions containing amplified genes lack a normal banding pattern, they appear homogeneous in a G-banded karyotype (see Fig. 7-28). The most interesting cases of amplification involve N-MYC in neuroblastoma and ERBB2 in breast cancers. N-MYC is amplified in 25% to 30% of neuroblastomas, and the amplification is associated with poor prognosis. In neuroblastomas with N-MYC amplification, the gene is present both in double minutes and homogeneous staining regions. ERBB2 amplification occurs in about 20% of breast cancers, and antibody therapy directed against this receptor has proven effective in this subset of tumors. Amplification of C-MYC, L-MYC, or N-MYC correlates with disease progression in small-cell cancer of the lung.
Epigenetic Changes
Epigenetics refers to reversible, heritable changes in gene expression that occur without mutation. Such changes involve post-translational modifications of histones and DNA methylation, both of which affect gene expression. In normal, differentiated cells, the majority of the genome is not expressed. Some portions of the genome are silenced by DNA methylation and histone modifications that lead to the compaction of DNA into heterochromatin. On the other hand, cancer cells are characterized by a global DNA hypomethylation and selective promoter-localized hypermethylation.140 Indeed, it has become evident during the past few years that tumor suppressor genes are sometimes silenced by hypermethylation of promoter sequences rather than mutation. One example is CDKN2A, a complex locus that encodes two tumor suppressors, p14/ARF and p16/INK4a from two different reading frames; p14/ARF is epigenetically silenced in colon and gastric cancers, while p16/INK4a is silenced in a wide variety of cancers. Since this locus produces two tumor suppressors which affect the p53 and Rb pathways, silencing this locus has the pleasing effect (from the cancer’s point of view) of removing two checkpoints with a single alteration. Other tumor suppressor genes subject to silencing by methylation include BRCA1 in breast cancer, VHL in renal cell carcinomas, and the MLH1 mismatch-repair gene in colorectal cancer.140 You will recall from Chapter 5 that methylation also participates in the phenomenon called genomic imprinting, in which the maternal or paternal allele of a gene or chromosome is modified by methylation and is inactivated. The reverse phenomenon—that is, demethylation of an imprinted gene leading to its biallelic expression (loss of imprinting)—can also occur in tumor cells.141 There has been great interest in developing potential therapeutic agents that act to demethylate DNA sequences in tumor suppressor genes. Recent data demonstrating that genomic hypomethylation causes chromosomal instability and induces tumors in mice greatly strengthen the notion that epigenetic changes may directly contribute to tumor development.141
The chromatin changes that contribute to carcinogenesis are less well understood. The current paradigm is that there is a histone code in which various modifications to the tails of histones, such as acetylation and methylation, lead to activation or repression of transcription. Several chromatin-modifying enzymes, such as EZH2, have been shown to be overexpressed in breast and prostate carcinomas.141 EZH2 is the enzymatic component of the multiprotein polycomb repressive complex 2, which places repressive chromatin marks at the promoter of genes. Although its targets in cancer in vivo have not yet been defined, in cell lines overexpression of EZH2 leads to the repression of tumor suppressors, such as p21. Interestingly, in flies and mammals the polycomb repressive complexes are required for the maintenance of stem cells, as well as to silence lineage-specific transcription factors until the proper cues signal differentiation. Inappropriate repression or expression of such genes could give cancer cells a stem cell–like, undifferentiated quality. There is, of course, significant cross-talk between the chromatin-remodeling enzymes and the DNA-methylation machinery. For example, the placement of repressive chromatin marks by enzymes like EZH2 in cancer cells results in the recruitment of DNA methylases, methylation of promoters, and durable repression of gene expression.
miRNAs and Cancer
As discussed in Chapter 5, miRNAs are small noncoding, single-stranded RNAs, approximately 22 nucleotides in length, that are incorporated into the RNA-induced silencing complex. The miRNAs mediate sequence-specific recognition of mRNAs and, through the action of the RNA-induced silencing complex, mediate post-transcriptional gene silencing. Given that miRNAs control cell growth, differentiation, and cell survival, it is not surprising that they play a role in carcinogenesis.142 miRNAs have been shown to undergo changes in expression in cancer cells, and frequent amplifications and deletions of miRNA loci have been identified in many cancers. As illustrated in Figure 7-39, miRNAs can participate in neoplastic transformation either by increasing the expression of oncogenes or by reducing the expression of tumor suppressor genes. If a miRNA inhibits the translation of an oncogene, a reduction in the quantity or function of that miRNA will lead to overproduction of the oncogene product; thus, the miRNA acts as a tumor suppressor. Conversely, if the target of a miRNA is a tumor suppressor gene, then overactivity of the miRNA can reduce the tumor suppressor protein; thus, the miRNA acts as an oncogene. Such relationships have already been established by miRNA profiling of several human tumors. For example, down-regulation or deletion of certain miRNAs in some leukemias and lymphomas results in increased expression of BCL2, the anti-apoptotic protein. Thus, by negatively regulating BCL2, such miRNAs behave as tumor suppressor genes. Similar miRNA-mediated upregulation of RAS and MYC oncogenes has also been detected in lung tumors and in certain B-cell leukemias, respectively. In some brain and breast tumors there is 5- to 100-fold greater expression of certain miRNAs. Although the targets of these miRNAs have not been identified, presumably they are tumor suppressor genes, whose activities are reduced by the overexpressed miRNA.
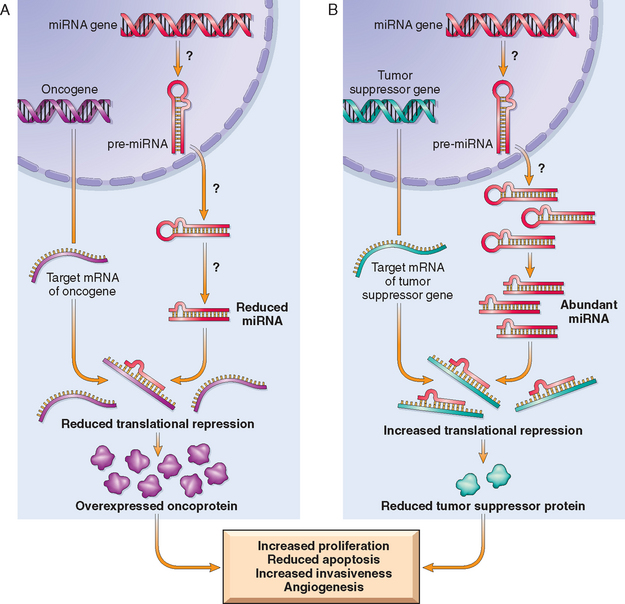
FIGURE 7-39 Role of miRNAs in tumorigenesis. A, Reduced activity of a miRNA that inhibits translation of an oncogene gives rise to an excess of oncoproteins. B, Overactivity of a miRNA that targets a tumor suppression gene reduces the production of the tumor suppressor protein. Question marks in A and B indicate that the mechanisms by which changes in the level or activity of miRNA are not entirely known.
These findings not only provide novel insights into carcinogenesis, they also have practical implications. For instance, drugs that inhibit or augment the functions of miRNAs could be useful in chemotherapy. Since miRNAs regulate normal cellular differentiation, the patterns of miRNA expression (“miRNA profiling”) can provide clues to the cell of origin and classification of tumors. Much remains to be learned about these oncogenic miRNAs, or so called “oncomirs.”