56 Individual variation and drug interaction
Overview
This chapter addresses sources of variation between individuals (interindividual variation) in their responses to drugs. Genetic variation in pharmacokinetic processes and pharmacodynamic response has been discussed in Chapter 11. In this chapter, we mention briefly some other important factors responsible for pharmacological variation, including age, pregnancy and disease, and describe in more detail the mechanisms underlying drug interaction (i.e. modfication of the action of one drug by another).
Introduction
Therapeutics would be a great deal easier if responses to the same dose of drug were always the same. In reality, inter- and even intraindividual variation is often substantial. Physicians need to be aware of the sources of such variation to prescribe drugs safely and effectively. Variation can be caused by different concentrations at sites of drug action or by different responses to the same drug concentration. The first kind is called pharmacokinetic variation and can occur because of differences in absorption, distribution, metabolism or excretion (Chs 8 and 9). The second kind is called pharmacodynamic variation.
Variation is usually quantitative in the sense that the drug produces a larger or smaller effect, or acts for a longer or shorter time, while still exerting qualitatively the same effect. In other cases, the action is qualitatively different. These are known as ‘idiosyncratic’ reactions (the Oxford English Dictionary defines idiosyncrasy as ‘the physical constitution peculiar to an individual or class’) and are often caused by genetic or immunological differences between individuals.
Factors Responsible For Quantitative Individual Variation
Ethnicity
Ethnic means ‘pertaining to race’, and many anthropologists are sceptical as to the value of this concept (see, for example, Cooper et al., 2003). Citizens of several modern societies are asked to define their race or ethnicity from a list of options (e.g. ‘white’, ‘black’, ‘mixed’, ‘Chinese’, ‘Asian’ or ‘other’ were the options provided by the UK Office of National Statistics for the 2001 National Census). Members of such self-defined groups share some characteristics on the basis of common genetic and cultural heritage, but there is obviously also enormous diversity within each group.
Despite the crudeness of such categorisation, it can give some pointers to drug responsiveness (Wood, 2001). One example is the evidence discussed in Chapter 22 that African-Americans with heart failure gain a mortality benefit from treatment with a combination of hydralazine plus a nitrate, whereas white Americans may not.
Some adverse effects may also be predicted on the basis of race; for example, many Chinese subjects differ from Europeans in the way that they metabolise ethanol, producing a higher plasma concentration of acetaldehyde, which can cause flushing and palpitations (Chs 48 and 57). Chinese subjects are considerably more sensitive to the cardiovascular effects of propranolol (Ch. 14) than white Europeans, whereas Afro-Caribbean individuals are less sensitive. Despite their increased sensitivity to β-adrenoceptor antagonists, Chinese subjects metabolise propranolol faster than white people, implying that the difference relates to pharmacodynamic differences in sensitivity at or beyond the β-adrenoceptors.
Overall effectiveness of gefitinib (Ch. 55) in treating patients with advanced lung tumours has been disappointing, but in about 10% of patients lung tumours shrink rapidly in response to this drug. Japanese patients are three times as likely as whites to respond in this way. The underlying difference is that patients who respond well have specific mutations in the receptor for epidermal growth factor (see Wadman, 2005). It is probable that many such ethnic differences are genetic in origin, but environmental factors, for example relating to distinctive dietary habits, may also contribute. It is important not to abandon the much more sophisticated search for ways to individualise medicine on the basis of pharmacogenomics (Ch. 11) just because the much simpler and cheaper process of asking patients to define their ethnic group has had some success: this should rather act as a spur. If such a crude and imperfect approach has had some success, think how much better we ought to be able to do with genomic testing!
Age
The main reason that age affects drug action is that drug elimination is less efficient in newborn babies and in old people, so that drugs commonly produce greater and more prolonged effects at the extremes of life. Other age-related factors, such as variations in pharmacodynamic sensitivity, are also important with some drugs. Physiological factors (e.g. altered cardiovascular reflexes) and pathological factors (e.g. hypothermia), which are common in elderly people, also influence drug effects. Body composition changes with age, fat contributing a greater proportion to body mass in the elderly, with consequent changes in distribution volume of drugs. Elderly people consume more drugs than do younger adults, so the potential for drug interactions (see below) is also increased. For fuller accounts of drug therapy in paediatrics and in the elderly, see, respectively, Fox & Balis (Ch. 23) and Abernethie (Ch. 24) in Atkinson et al., 2006.
Effect of Age on Renal Excretion of Drugs
Glomerular filtration rate (GFR) in the newborn, normalised to body surface area, is only about 20% of the adult value, and tubular function is also less. Accordingly, plasma elimination half-lives of renally eliminated drugs are longer in neonates than in adults (Table 56.1). In babies born at term, renal function increases to values similar to those in young adults in less than a week, and continues to increase to a maximum of approximately twice the adult value at 6 months of age. Improvement in renal function occurs more slowly in premature infants. Renal immaturity in premature infants can have a substantial effect on drug elimination. For example, in premature newborn babies, the antibiotic gentamicin has a plasma half-life of ≥ 18 h, compared with 1–4 h for adults and approximately 10 h for babies born at term. It is therefore necessary to reduce and/or space out doses to avoid toxicity in premature babies.
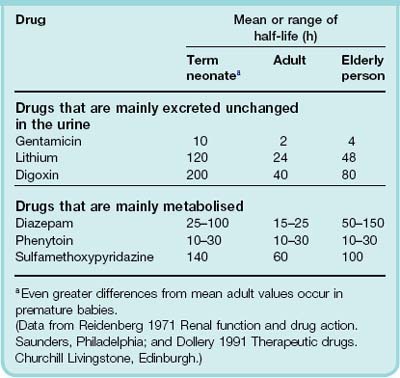
Glomerular filtration rate declines slowly from about 20 years of age, falling by about 25% at 50 years and by 50% at 75 years. Figure 56.1 shows that the renal clearance of digoxin in young and old subjects is closely correlated with creatinine clearance, a measure of GFR. Consequently, chronic administration over the years of the same daily dose of digoxin to an individual as he or she ages leads to a progressive increase in plasma concentration, and this is a common cause of glycoside toxicity in elderly people (see Ch. 21).
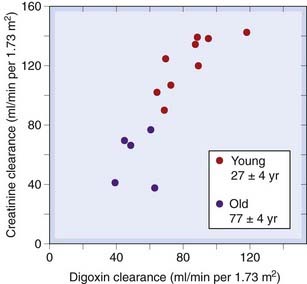
Fig. 56.1 Relationship between renal function (measured as creatinine clearance) and digoxin clearance in young and old subjects.
(From Ewy G A et al. 1969 Circulation 34: 452.)
 The age-related decline in GFR is not reflected by an increase in plasma creatinine concentration, as distinct from creatinine clearance. Plasma creatinine typically remains within the normal adult range in elderly persons despite substantially diminished GFR. This is because creatinine synthesis is reduced in elderly persons because of their reduced muscle mass. Consequently, a ‘normal’ plasma creatinine in an elderly person does not indicate that they have a normal GFR. Failure to recognise this and reduce the dose of drugs that are eliminated by renal excretion can lead to drug toxicity.
The age-related decline in GFR is not reflected by an increase in plasma creatinine concentration, as distinct from creatinine clearance. Plasma creatinine typically remains within the normal adult range in elderly persons despite substantially diminished GFR. This is because creatinine synthesis is reduced in elderly persons because of their reduced muscle mass. Consequently, a ‘normal’ plasma creatinine in an elderly person does not indicate that they have a normal GFR. Failure to recognise this and reduce the dose of drugs that are eliminated by renal excretion can lead to drug toxicity.
Effect of Age on Drug Metabolism
Several important enzymes, including hepatic microsomal oxidase, glucuronyltransferase, acetyltransferase and plasma esterases, have low activity in neonates, especially if premature. These enzymes take 8 weeks or longer to reach the adult level of activity. The relative lack of conjugating activity in the newborn can have serious consequences, as in kernicterus caused by drug displacement of bilirubin from its binding sites on albumin (see below) and in the ‘grey baby’ syndrome caused by the antibiotic chloramphenicol (see Ch. 50). This sometimes fatal condition, at first thought to be a specific biochemical sensitivity to the drug in young babies, actually results simply from accumulation of very high tissue concentrations of chloramphenicol because of slow hepatic conjugation. Chloramphenicol is no more toxic to babies than to adults provided the dose is reduced to make allowance for this. Slow conjugation is also one reason why morphine (which is excreted mainly as the glucuronide, see Ch. 41) is not used as an analgesic in labour, because drug transferred via the placenta has a long half-life in the newborn baby and can cause prolonged respiratory depression.
The activity of hepatic microsomal enzymes declines slowly (and very variably) with age, and the distribution volume of lipid-soluble drugs increases, because the proportion of the body that is fat increases with advancing age. The increasing half-life of the anxiolytic drug diazepam with advancing age (Fig. 56.2) is one consequence of this. Some other benzodiazepines and their active metabolites show even greater age-related increases in half-life. Because half-life determines the time course of drug accumulation during repeated dosing (Ch. 10), insidious effects, developing over days or weeks, can occur in elderly people and may be misattributed to age-related memory impairment rather than to drug accumulation. The effect of age is less marked for many other drugs, but even though the mean half-life may not change much, there is often a striking increase in the variability of half-life between individuals with increasing age. This is important, because a population of old people will contain some individuals with grossly reduced rates of drug metabolism, whereas such extremes do not occur so commonly in young adult populations. Drug regulatory authorities therefore usually require studies in elderly patients as part of drug evaluation.
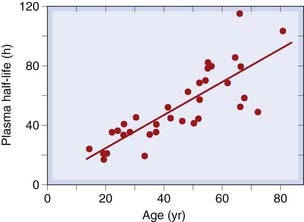
Age-Related Variation in Sensitivity to Drugs
The same plasma concentration of a drug can cause different effects in young and old subjects. Benzodiazepines (Ch. 43) exemplify this, producing more confusion and less sedation in elderly than in young subjects; similarly, hypotensive drugs (Ch. 22) cause postural hypotension more commonly in elderly than in younger adult patients.
Pregnancy
Pregnancy causes physiological changes that influence drug disposition (Ch. 8) in mother and fetus. Maternal plasma albumin concentration is reduced, influencing drug protein binding. Cardiac output is increased, leading to increased renal blood flow and GFR, and increased renal elimination of drugs. Lipophilic molecules rapidly traverse the placental barrier, whereas transfer of hydrophobic drugs is slow, limiting fetal drug exposure following a single maternal dose. The placental barrier excludes some drugs (e.g. low-molecular-weight heparins; Ch. 24) so effectively that they can be administered chronically to the mother without causing effects in the fetus. However, drugs that are transferred to the fetus are eliminated more slowly than from the mother. The activity of most drug-metabolising enzymes in fetal liver is much less than in the adult. Furthermore, the fetal kidney is not an efficient route of elimination because excreted drug enters the amniotic fluid, which is swallowed by the fetus. For a fuller account, see Striker & Frederiksen (Ch. 22) in Atkinson et al., 2006.
Disease
Therapeutic drugs are prescribed to patients, so effects of disease on drug response are very important in clinical pharmacology. Detailed consideration is beyond the scope of this book, and interested readers should refer to a clinical text such as the chapters on renal and hepatic disease in Atkinson et al., 2006. Disease can cause pharmacokinetic or pharmacodynamic variation. Common disorders such as impaired renal or hepatic function predispose to toxicity by causing unexpectedly intense or prolonged drug effects as a result of increased drug concentration following a standard dose. Drug absorption is slowed in conditions causing gastric stasis (e.g. migraine, diabetic neuropathy) and may be incomplete in patients with malabsorption owing to ileal or pancreatic disease or to oedema of the ileal mucosa caused by heart failure or nephrotic syndrome. Nephrotic syndrome (characterised by heavy proteinuria, oedema and a reduced concentration of albumin in plasma) alters drug absorption because of oedema of intestinal mucosa; alters drug disposition through changes in binding to plasma albumin; and causes insensitivity to diuretics such as furosemide that act on ion transport mechanisms on the lumenal surface of tubular epithelium (Ch. 28), through binding to albumin in tubular fluid. Hypothyroidism is associated with increased sensitivity to several widely used drugs (e.g. pethidine), for reasons that are poorly understood. Hypothermia (to which elderly persons, in particular, are predisposed) markedly reduces the clearance of many drugs.
Other disorders affecting receptors and signal transduction mechanisms (see Ch. 3), although uncommon, illustrate mechanisms that may prove to be of more general applicability. Examples include:
Genetic factors (see Ch. 11)
Idiosyncratic Reactions
An idiosyncratic reaction is a qualitatively abnormal, and usually harmful, drug effect that occurs in a small proportion of individuals. For example, chloramphenicol causes aplastic anaemia in approximately 1 in 50 000 patients (Ch. 50). In many cases, genetic anomalies are responsible. Glucose 6-phosphate dehydrogenase (G6PD) deficiency and the hepatic porphyrias are well-understood examples of this (Ch. 11). Malignant hyperthermia is a metabolic reaction to drugs including suxamethonium and various inhalational anaesthetics and antipsychotic drugs. Susceptibility to these drugs in affected individuals is caused by an inherited abnormality in the Ca2+ release channel known as the ryanodine receptor located in the sarcoplasmic reticulum of striated muscle (Ch. 4).
Immunological mechanisms underlie many idiosyncratic reactions. Propensity to these is genetically determined (Ch. 11).They are considered further in Chapter 57.
Drug Interactions
Many patients, especially elderly ones, are treated continuously with one or more drugs for chronic diseases such as hypertension, heart failure, osteoarthritis and so on. Acute events (e.g. infections, myocardial infarction) are treated with additional drugs. The potential for drug interactions is therefore substantial, and drug interactions account for 5–20% of adverse drug reaction. These may be serious (approximately 30% of fatal adverse drug reactions are estimated to be the consequence of drug interaction) and may be misattributed to the natural history of disease (e.g. rejection of a transplanted kidney may be attributed to this when it was actually caused by loss of effectiveness of immunosuppressant medication as a result of drug interaction; see below). Drugs can also interact with chemical entities in other dietary constituents (e.g. grapefruit juice, which downregulates expression of CYP3A4 in the gut) and herbal remedies (such as St John’s wort; Ch. 46). The administration of one chemical entity (A) can alter the action of another (B) by one of two general mechanisms:1
For such interactions to be important clinically, it is necessary that the therapeutic range of drug B is narrow (i.e. that a small reduction in effect will lead to loss of efficacy and/or a small increase in effect will lead to toxicity). For pharmacokinetic interactions to be clinically important, it is also necessary that the concentration–response curve of drug B is steep (so that a small change in plasma concentration leads to a substantial change in effect). For many drugs, these conditions are not met: even quite large changes in plasma concentrations of relatively non-toxic drugs such as penicillin are unlikely to give rise to clinical problems, because there is usually a comfortable safety margin between plasma concentrations produced by usual doses and those resulting either in toxicity or in loss of efficacy. Several drugs do have steep concentration–response relationships and a narrow therapeutic margin and, for these, drug interactions can cause major problems, for example with antithrombotic, antidysrhythmic, antiviral and antiepileptic drugs; lithium; and several antineoplastic and immunosuppressant drugs.
Pharmacodynamic Interaction
Pharmacodynamic interaction can occur in many different ways (including those discussed under Drug antagonism in Ch. 2). There are many mechanisms, and some examples of practical importance are probably more useful than attempts at classification:
Pharmacokinetic Interaction
All the four major processes that determine pharmacokinetics—absorption, distribution, metabolism and excretion—can be affected by drugs. Some of the more important mechanisms are given here, with examples.
Absorption
Gastrointestinal absorption is slowed by drugs that inhibit gastric emptying, such as atropine or opiates, or accelerated by drugs that hasten gastric emptying (e.g. metoclopramide; see Ch. 29). Alternatively, drug A may interact with drug B in the gut in such a way as to inhibit absorption of B (cf. pharmaceutical interactions; see footnote 1). For example, Ca2+ or Fe2+ each form insoluble complexes with tetracycline that retard its absorption; colestyramine, a bile acid-binding resin, binds several drugs (e.g. warfarin, digoxin), preventing their absorption if administered simultaneously. Another example is the addition of adrenaline (epinephrine) to local anaesthetic injections; the resulting vasoconstriction slows the absorption of the anaesthetic, thus prolonging its local effect (Ch. 42).
Drug Distribution
One drug may alter the distribution of another, by competing for a common binding site on plasma albumen or tissue protein, but such interactions are seldom clinically important unless accompanied by a separate effect on drug elimination (see below). Displacement of a drug from binding sites in plasma or tissues transiently increases the concentration of free (unbound) drug, but this is followed by increased elimination, so a new steady state results in which total drug concentration in plasma is reduced but the free drug concentration is similar to that before introduction of the second ‘displacing’ drug. Consequences of potential clinical importance are as follow:
Although many drugs have appreciable affinity for plasma albumin (Ch. 8), and therefore might potentially be expected to interact in these ways, there are rather few instances of clinically important interactions of this type. Protein-bound drugs that are given in large enough dosage to act as displacing agents include various sulfonamides and chloral hydrate; trichloracetic acid, a metabolite of chloral hydrate, binds very strongly to plasma albumin. Displacement of bilirubin from albumin by such drugs in jaundiced premature neonates can have clinically disastrous consequences: bilirubin metabolism is undeveloped in the premature liver, and unbound bilirubin can cross the immature blood–brain barrier and cause kernicterus (staining of the basal ganglia by bilirubin). This causes a distressing and permanent disturbance of movement known as choreoathetosis, characterised by involuntary writhing and twisting movements in the child.
Phenytoin dose is adjusted according to measurement of its concentration in plasma, and such measurements do not routinely distinguish bound from free phenytoin (that is, they reflect the total concentration of drug). Introduction of a displacing drug in an epileptic patient whose condition is stabilised on phenytoin (Ch. 44) reduces the total plasma phenytoin concentration owing to increased elimination of free drug, but there is no loss of efficacy because the concentration of unbound (active) phenytoin at the new steady state is unaltered. If it is not appreciated that the therapeutic range of plasma concentrations has been reduced in this way, an increased dose may be prescribed, resulting in toxicity.
There are several instances where drugs that alter protein binding additionally reduce elimination of the displaced drug, causing clinically important interactions. Phenylbutazone displaces warfarin from binding sites on albumin, and more importantly selectively inhibits metabolism of the pharmacologically active (S) isomer (see below), prolonging prothrombin time and resulting in increased bleeding (Ch. 24). Salicylates displace methotrexate from binding sites on albumin and reduce its secretion into the nephron by competition with the organic anion transporter (OAT; Ch. 9). Quinidine and several other antidysrhythmic drugs including verapamil and amiodarone (Ch. 21) displace digoxin from tissue-binding sites while simultaneously reducing its renal excretion; they consequently can cause severe dysrhythmias through digoxin toxicity.
Drug Metabolism
Drugs can either induce (Table 56.2) or inhibit (Table 56.3) drug-metabolising enzymes.
Table 56.2 Examples of drugs that induce drug-metabolising enzymes

Table 56.3 Examples of drugs that inhibit drug-metabolising enzymes
| Drugs inhibiting enzyme action | Drugs with metabolism affected |
|---|---|
| Allopurinol | Mercaptopurine, azathioprine |
| Chloramphenicol | Phenytoin |
| Cimetidine | Amiodarone, phenytoin, pethidine |
| Ciprofloxacin | Theophylline |
| Corticosteroids | Tricyclic antidepressants, cyclophosphamide |
| Disulfiram | Warfarin |
| Erythromycin | Ciclosporin, theophylline |
| Monoamine oxidase inhibitors | Pethidine |
| Ritonavir | Saquinavir |
Enzyme induction
Enzyme induction (e.g. by anticonvulsants, ethanol or rifampicin; see Ch. 9) is an important cause of drug interaction. The slow onset of induction and slow recovery after withdrawal of the inducing agent together with the potential for selective induction of one or more CYP isoenzymes contributes to the insidious nature of the clinical problems that induction presents. Adverse clinical outcomes from such interactions are very diverse including graft rejection as a result of loss of effectiveness of immunosuppressive treatment, seizures due to loss of anticonvulsant effectiveness, unwanted pregnancy and thrombosis (from loss of effectiveness of warfarin) or bleeding (from failure to recognise the need to reduce warfarin dose when induction wanes). Over 200 drugs cause enzyme induction and thereby decrease the pharmacological activity of a range of other drugs. Some examples are given in Table 56.2. Because the inducing agent is often itself a substrate for the induced enzymes, the process can result in slowly developing tolerance. This pharmacokinetic kind of tolerance is generally less marked than pharmacodynamic tolerance, for example to opioids (Ch. 41), but it is clinically important when starting treatment with carbamazepine (Ch. 44). This is initiated at a low dose to avoid toxicity (because liver enzymes are not induced initially) and gradually increased over a period of a few weeks, during which it induces its own metabolism.
Figure 56.3 shows how the antibiotic rifampicin, given for 3 days, reduces the effectiveness of warfarin as an anticoagulant. Conversely, enzyme induction can increase toxicity of a second drug if the toxic effects are mediated via an active metabolite. Paracetamol (acetaminophen) toxicity is a case in point (see Fig. 57.1): this is caused by its CYP metabolite N-acetyl-p-benzoquinone imine. Consequently, the risk of serious hepatic injury following paracetamol overdose is increased in patients in whom CYP has been induced, for example by chronic alcohol consumption. Variability in rates of drug metabolism between individuals results partly from varying exposure to environmental chemicals, some of which are powerful enzyme inducers.
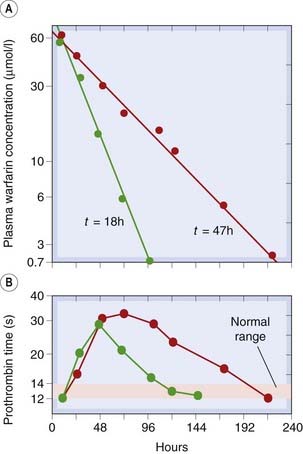
Fig. 56.3 Effect of rifampicin on the metabolism and anticoagulant action of warfarin.
[A] Plasma concentration of warfarin (log scale) as a function of time following a single oral dose of 5 µmol/kg body weight. After the subject was given rifampicin (600 mg daily for a few days), the plasma half-life of warfarin decreased from 47 h (red curve) to 18 h (green curve). [B] The effect of a single dose of warfarin on prothrombin time under normal conditions (red curve) and after rifampicin administration (green curve).
(Redrawn from O’Reilly 1974 Ann Intern Med 81: 337.)
Enzyme induction is exploited therapeutically by administering phenobarbital to premature babies to induce glucuronyltransferase, thereby increasing bilirubin conjugation and reducing the risk of kernicterus (see above).
Enzyme inhibition
Enzyme inhibition, particularly of CYP enzymes, slows the metabolism and hence increases the action of other drugs inactivated by the enzyme. Such effects can be clinically important and are major considerations in the treatment of patients with HIV infection with triple and quadruple therapy, because several protease inhibitors are potent CYP inhibitors (Ch. 51). Other examples of drugs that are enzyme inhibitors are shown in Table 56.3. To make life even more difficult, several inhibitors of drug metabolism influence the metabolism of different stereoisomers selectively. Examples of drugs that inhibit the metabolism of the active (S) and less active (R) isomers of warfarin in this way are shown in Table 56.4.
Table 56.4 Stereoselective and non-stereoselective inhibition of warfarin metabolism
| Inhibition of metabolism | Drug(s) |
|---|---|
| Stereoselective for (S) isomer | Phenylbutazone Metronidazole Sulfinpyrazone Trimethoprim–sulfamethoxazole Disulfiram |
| Stereoselective for (R) isomer | Cimetidinea Omeprazolea |
| Non-stereoselective effect on both isomers | Amiodarone |
a Minor effect only on prothrombin time.
From Hirsh 1991 N Engl J Med 324: 1865–1875.
The therapeutic effects of some drugs are a direct consequence of enzyme inhibition (e.g. the xanthine oxidase inhibitor allopurinol, used to prevent gout; Ch. 26). Xanthine oxidase metabolises several cytotoxic and immunosuppressant drugs, including mercaptopurine (the active metabolite of azathioprine), the action of which is thus potentiated and prolonged by allopurinol. Disulfiram, an inhibitor of aldehyde dehydrogenase used to produce an aversive reaction to ethanol (see Ch. 48), also inhibits metabolism of other drugs, including warfarin, which it potentiates. Metronidazole, an antimicrobial used to treat anaerobic bacterial infections and several protozoal diseases (Chs 50 and 53), also inhibits this enzyme, and patients prescribed it are advised to avoid alcohol for this reason.
In other instances, inhibition of drug metabolism is less expected because enzyme inhibition is not the main mechanism of action of the offending agents. Thus, glucocorticosteroids and cimetidine enhance the actions of a range of drugs including some antidepressant and cytotoxic drugs.
When a drug works through an active metabolite, inhibition of its metabolism can result in loss of activity. An example of topical concern is an interaction between proton pump inhibitors (such as omeprazole, Ch. 29) and the antiplatelet drug clopidogrel (Ch. 24). These have been widely co-prescribed (because clopidogrel is often used with other antithrombotic drugs so there is a high risk of bleeding from the stomach—omeprazole reduces this). Clopidogrel works through an active metabolite formed by CYP2C19 (people who carry a genetic variant where CYP2C19 is less active have an increased risk of thrombosis during treatment with clopidogrel). Omeprazole inhibits CYP2C19, and also reduces the antiplatelet action of clopidogrel. It is not yet clear how clinically important this may be, but the Food and Drug Administration has warned against concomitant use of these drugs (http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm190848.htm). It is anticipated that other enzyme inhibitors will have a similar deleterious effect.
As with induction, interactions caused by enzyme inhibition are hard to anticipate from first principles. If in doubt about the possibility of an interaction, it is best to look it up (e.g. in the British National Formulary, which has an invaluable appendix on drug interactions indicating which are of known clinical importance).
Haemodynamic effects
Variations in hepatic blood flow influence the rate of inactivation of drugs that are subject to extensive presystemic hepatic metabolism (e.g. lidocaine, propranolol). A reduced cardiac output reduces hepatic blood flow, so drugs that reduce cardiac output (e.g. propranolol) reduce the rate of metabolism of lidocaine by this mechanism. Extraction of lidocaine by liver approaches 100% and measurement of lidocaine clearance has been used to estimate hepatic blood flow in the same way as clearance of p-aminohippuric acid (PAH) has been used to estimate renal blood flow (Ch. 9).
Drug Excretion
The main mechanisms by which one drug can affect the rate of renal excretion of another are by:
Inhibition of tubular secretion
Probenecid (Ch. 28) was developed to inhibit penicillin secretion and thus prolong its action. It also inhibits the excretion of other drugs, including zidovudine (see Ch. 51). Other drugs have an incidental probenecid-like effect and can enhance the actions of substances that rely on tubular secretion for their elimination. Table 56.5 gives some examples. Because diuretics act from within the tubular lumen, drugs that inhibit their secretion into the tubular fluid, such as NSAIDs, reduce their effect.
Table 56.5 Examples of drugs that inhibit renal tubular secretion
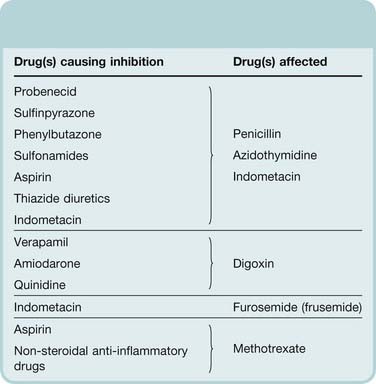
Alteration of urine flow and pH
Diuretics tend to increase the urinary excretion of other drugs and their metabolites, but this is seldom immediately clinically important. Conversely, loop and thiazide diuretics indirectly increase the proximal tubular reabsorption of lithium (which is handled in a similar way as Na+), and this can cause lithium toxicity in patients treated with lithium carbonate for mood disorders (Ch. 46). The effect of urinary pH on the excretion of weak acids and bases is put to use in the treatment of poisoning with salicylate (see Ch. 8), but is not a cause of accidental interactions.
References and Further Reading
Barry M., Mulcahy F., Merry C., et al. Pharmacokinetics and potential interactions amongst antiretroviral agents used to treat patients with HIV infection. Clin. Pharmacokinet.. 1999;36:289-304. (Multidrug combinations have transformed the outlook for patients with HIV infection; drug interactions are one of the main problems associated with these)
Carmichael D.J.S. Handling of drugs in kidney disease. In: Davison A.M., Cameron J.S., Grunfeld J.P., et al, editors. Oxford textbook of clinical nephrology. third ed. Oxford: Oxford University Press; 2005:2599-2618. (Principles and practice of dose adjustment in patients with renal failure)
Cooper R.S., Kaufman J.S., Ward R. Race and genomics. N. Engl. J. Med.. 2003;348:1166-1170. (Scholarly and appropriately sceptical analysis)
Fugh-Berman A., Ernst E. Herb–drug interactions: review and assessment of report reliability. Br. J. Clin. Pharmacol.. 2001;52:587-595. (Warfarin the most common drug, St John’s wort the most common herb; more data needed! See also Fugh-Berman, A., 2000. Lancet 355, 134–138)
Hanratty C.G., McGlinchey P., Johnston G.D., Passmore A.P. Differential pharmacokinetics of digoxin in elderly patients. Drugs Aging. 2000;17:353-362. (Reviews pharmacokinetics of digoxin in relation to age, concomitant disease and interacting drugs)
Lin J.H., Liu A.Y.H. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu. Rev. Pharmacol. Toxicol.. 2001;41:535-567. (Examines sources of interindividual variability in inhibition and induction of P450 enzymes)
Rowland M., Tozer T.N. Clinical pharmacokinetics and pharmacodynamics. Concepts and applications. Baltimore: Wolters Kluwer/ Lippincott, Williams & Wilkins; 2010. (Online simulations by H Derendorf and G Hochhaus)
Sproule B.A., Hardy B.G., Shulman K.I. Differential pharmacokinetics in elderly patients. Drugs Aging. 2000;16:165-177. (Reviews age-related changes in pharmacodynamics as well as pharmacokinetics and drug interactions, all of which are clinically important)
Westphal J.F. Macrolide-induced clinically relevant drug interactions with cytochrome P450A (CYP) 3A4: an update focused on clarithromycin, azithromycin and dirithromycin. Br. J. Clin. Pharmacol.. 2000;50:285-295. (Review: theophylline, ciclosporine, warfarin, involvement of P-glycoprotein as well as metabolism)
Xie H.-G., Kim R.B., Wood A.J.J., Stein C.M. Molecular basis of ethnic differences in drug disposition and response. Annu. Rev. Pharmacol. Toxicol.. 2001;41:815-850. (Recent developments in understanding genetic variations that may underlie ethnic differences in drug-metabolising enzymes, transporters, receptors and second messenger systems)
Zevin S., Benowitz N.L. Drug interactions with tobacco smoking—an update. Clin. Pharmacokinet.. 1999;36:425-438. (Polycyclic aromatic hydrocarbons in tobacco smoke induce various P450 enzymes. ‘Cigarette smoking should be specifically studied in clinical trials of new drugs’)
Atkinson A.J.Jr, Abernethie D.R., Daniels C.E., et al. Principles of clinical pharmacology, second ed. San Diego: Academic Press; 2006. (Includes detailed accounts of clinical aspects including effects of renal and liver disease on pharmacokinetics, of effects of age and on drug therapy in pregnant and nursing women)
Wadman M. Drug targeting: is race enough? Nature. 2005;435:1008-1009. (No!)
Wood A.J.J. Racial differences in response to drugs—pointers to genetic differences. N. Engl. J. Med.. 2001;344:1393-1396.
1A third category of pharmaceutical interactions should be mentioned, in which drugs interact in vitro so that one or both are inactivated. No pharmacological principles are involved, just chemistry. An example is the formation of a complex between thiopental and suxamethonium, which must not be mixed in the same syringe. Heparin is highly charged and interacts in this way with many basic drugs; it is sometimes used to keep intravenous lines or cannulae open and can inactivate basic drugs if they are injected without first clearing the line with saline.
2The interaction with diuretics may involve a pharmacokinetic interaction in addition to the pharmacodynamic effect described here, because NSAIDs compete with weak acids, including diuretics, for renal tubular secretion; see below.