Bone Marrow, Blood Cells, and Lymphatic System
Canine Distemper (Morbillivirus; Enveloped RNA Virus): See the section on the Nervous System.
Equine Infectious Anemia (Equine Infectious Anemia Virus; Enveloped RNA Virus): The mechanism of injury in equine infectious anemia is inflammation (and proliferation [hypertrophy and hyperplasia]) of the monocyte-macrophage and lymphoid systems, particularly in the spleen and lymph nodes, resulting in chronic-active splenitis and lymphadenitis. Virus does not cause cell death. Gross lesions include an enlarged spleen (splenomegaly) and lymph nodes (lymphadenomegaly) with abundant white-gray lymphoid tissue arranged in follicles and solid sheets of cells that often bulge from cut surfaces.
Horses initially encounter virus through penetrating wounds of the blood vascular system, either from fly or mosquito bites or needles. In the blood, virus infects monocytes, but because they are not fully differentiated macrophages, virus cannot fully replicate. Thus monocytes spread virus via leukocyte trafficking in the circulatory system to all organ systems and migrate through blood vessel walls. After monocytes enter tissue, they differentiate into tissue macrophages and virus can replicate in these cells and serve to infect other macrophages and lymphocytes, especially in lymphoid tissues such as spleen and lymph nodes. Infected macrophages produce proinflammatory chemokines and cytokines that recruit additional monocytes and lymphocytes into organs, thus splenomegaly and lymphadenomegaly ensue. Virus does not cause cell death. It appears virus expresses an envelope glycoprotein (gp90 and likely other proteins) that attach and bind to equine lentivirus receptor-1 present in cell membranes of monocytes and macrophages.
The two clinical phases of equine infectious anemia are acute and chronic. In the acute phase, horses have recurring fever, anemia, thrombocytopenia, and petechia with interspersed periods of quiescence. Fever is likely attributable to release of proinflammatory cytokines and endogenous pyrogens from activated macrophages during the leukocyte trafficking stages of the disease. Anemia occurs from phagocytosis and complement-mediated lysis of erythrocytes that have had their membranes altered by virus, antibody, complement, and/or fibrinogen. Pulmonary intravascular macrophages, Kupffer cells, and fixed macrophages lining vascular sinusoids in the spleen and lymph nodes are reservoirs for virus and release it into the bloodstream. Cell-free virus adsorbs onto the surface of red blood cells (and likely platelets) in the circulatory system. Adsorbed viral proteins act as haptens that are recognized as foreign by cells of the monocyte-macrophage system and are phagocytosed. Additionally, the hapten is processed and presented to lymphocytes, leading to a humoral immune response and the generation of plasma cells that secrete antibody against the hapten and other antigens on the red blood cell membrane (type II hypersensitivity response). If the hapten-antibody complex fixes complement, red blood cells are lysed intravascularly. If complement is not fixed, red blood cells are phagocytosed by cells of the monocyte-macrophage system and lysed extravascularly. Both of these mechanisms result in severe anemia. The cause of thrombocytopenia is less clear and is thought to occur because of activation of platelets and concurrent binding of fibrinogen to the surface of platelets during acute viremic phases of the disease. It is likely that activated platelets are quickly phagocytosed by the monocyte-macrophage system leading to thrombocytopenia. Petechial hemorrhages may be attributable to vascular injury caused by direct infection of endothelial cells by virus or more likely by a secondary response to injury induced by innate and adaptive defense mechanisms.
In chronic equine infectious anemia, recurrence of disease is caused by antigenic variation of surface glycoproteins of the virus. This genetic variation results in virus that expresses new surface glycoproteins, thus beginning anew the process of developing effective cell-mediated and humoral responses. Long-lasting control of equine infectious anemia appears to require that the adaptive immune response controls the disease before antigenic variation occurs. Large quantities of virus are replicated in cells of the monocyte-macrophage system, and virus is not eliminated from these cells during the acute phase of the disease. As adaptive immune responses develop, cytotoxic T lymphocytes are thought to control a limited extent viremia and virus replication in infected monocytes and macrophages. However, it appears that control of the disease (no or minimal anemia and thrombocytopenia) is linked to an effective antibody response against the virus that takes 6 to 8 months to develop.
Enzootic Bovine Lymphoma (Lymphosarcoma, Bovine Leukosis Virus-Associated Malignant Lymphoma, Deltaretrovirus: Bovine Leukemia Virus, Enveloped RNA Virus): The mechanism of injury in enzootic bovine lymphoma is provirus-induced malignant transformation of B lymphocytes. Gross lesions include proliferation of neoplastic cells and their infiltration into perivascular spaces in organ systems resulting in (1) generalized enlargement of affected organs with increased pallor or (2) the formation of one or more solid white nodules distributed at random in the affected tissue (see Figs. 7-81, 10-54, 10-55, 13-56, B, 13-80, and 13-86). Additionally, cells can occupy and proliferate in confined spaces causing compressive atrophy of tissue in these spaces such as axons in the spinal cord, hemopoietic cells in the bone marrow, and the retina in the eye. Organ systems commonly having lesions include superficial and visceral lymph nodes and thymus, skin, abomasum, heart, spleen, kidneys, uterus (caruncles), spinal meninges, retrobulbar lymphatic tissue, bone, and bone marrow. Malignant transformation is a sequence of events in which normal cells acquire the biologic behaviors of neoplastic cells such as uncontrolled growth, tissue invasion, and metastasis. In cattle, it takes several years for this transformation to occur and be manifested in overt lymphoma. This long prodromal period is likely caused by the complexity and interplay of injurious and reparative processes induced by the provirus that eventually result in dysfunction or mutation of regulatory cell cycle genes. Bovine leukemia virus infects B lymphocytes and thus is not free as a virus in blood or body fluids but is a provirus, cell-associated, and integrated into the host cell’s genome. When its replication cycle is completed, new virus is released from provirus-infected B lymphocytes. New virus serves to sustain and amplify the infection by infecting naïve B lymphocytes and cells of the monocyte-macrophage system.
Cattle and calves encounter provirus-infected B lymphocytes in blood, inflammatory exudates, and colostrum or milk. Provirus-infected B lymphocytes must gain access to the blood vascular and/or lymphatic systems and eventually to tissues and target cells suitable for infection. When it gains access to tissues, it is unclear whether B lymphocytes (1) behave as trafficking leukocytes and migrate into the vascular and/or lymphatic systems to spread new virus to other cells and tissues or (2) undergo cytolysis and release virus into tissues to infect local tissue macrophages, lymphocytes, or dendritic cells such as Langerhans’ cells. The use of needles or surgical instruments contaminated with blood (provirus-infected B lymphocytes) can transfer these cells directly into the vascular system or place them in vascularized subcutaneous tissues or muscle in close proximity to capillary and lymphatic vascular beds. Such exposure may require traumatic injury to skin or mucous membranes. Insect bites can apparently result in the same outcome. In either case, provirus-infected B lymphocytes deposited in these locations encounter cells of the monocyte-macrophage and lymphoid systems and dendritic cells. It has been shown that virus can infect these cells, but whether these cells spread virus or provirus to regional lymph nodes and then systemically via leukocyte trafficking in these cells or in B lymphocytes is unknown.
Transplacental spread of bovine leukemia virus from cows to calves also occurs through the blood. Provirus-infected B lymphocytes can also be present in inflammatory exudates, such as those occurring with postpartum metritis or vaginitis, and must gain access to capillary and lymphatic beds in host animals as previously described. Finally, provirus-infected B lymphocytes can be present in colostrum or milk, and it has been suggested that enzootic bovine lymphoma can result from virus entering the body via the alimentary system and gaining access to the blood vascular system. However, the role, as examples, of alimentary peristalsis, gastric acidity, mucosal mucus barrier systems, mucosal epithelial barrier systems, mucosal immunity, and M cells has not been adequately addressed in experimental studies.
It appears that by whatever route used by provirus-infected B lymphocytes to enter the body, it must gain access to the blood vascular system to establish, sustain, and amplify an infection regionally and systemically. Although hypothetical, provirus-infected B lymphocytes could behave as typical trafficking leukocytes and thereby attach to and migrate through mucosae of the oral and nasal pharynx and gain access to local MALTs, lymphatic vessels, regional lymph nodes, and systemic lymphoid tissues. Virus uses bovine leukemia virus envelope glycoproteins (gp51, gp30) to attach to and enter naïve host B lymphocytes that express a novel membrane protein called bovine leukemia virus–binding receptor. Other studies have shown that B lymphocytes expressing surface immunoglobulin M and cell surface markers CD5 and CD11b are more susceptible to infection with virus; however, the role of these molecules as receptors is unclear.
The mechanism of B lymphocyte transformation has not been established. Transformation may be linked to a mechanism called gene transactivation. When the genome of bovine leukemia virus (provirus) is integrated into the genome of a B lymphocyte, the provirus asserts control over the transcriptional and translational organelles and processes of the host cell. Genes of the bovine leukemia provirus express a protein called bovine leukemia virus Tax protein (p34tax) that appears to stimulate the proliferation (increased mitoses) of B lymphocytes and increases viral replication in host cells. Tax protein also interacts with host cell genes and appears to transactivate genes that express proteins modulating host cell growth, such as cell division and differentiation, and are involved in regulatory steps of cell proliferation and longevity. Experimentally, Tax protein has been shown to be able to immortalize rat embryo fibroblasts in tissue culture and to cooperate with an oncogene to transform tissue culture cells that can then be grown as tumors in live animals. Collectively, these findings suggest that transformation of B lymphocytes, leading to bovine lymphoma, is linked to the likely long-term actions of p34tax on host cell regulatory genes, but the chronologic stages of transformation are uncertain. Studies suggest that transformation may also result from Tax protein forming complexes with proteins expressed by tumor-suppressor genes such as p53, whereas other studies suggest that point mutations in the p53 gene is one of the critical events leading to lymphoma. The proteins translated from tumor-suppressor genes have an inhibitory effect on the regulation of the cell cycle and function to inhibit cell division, inhibit division of cells with damaged DNA, initiate apoptosis of cells with damaged DNA, and amplify cell adhesion (metastasis suppressors). When the activities of the p53 gene and its protein gene products are perturbed or inhibited, transformation of affected cells could occur.
Feline Leukemia (Feline Leukemia Virus, Retrovirus, Enveloped RNA Virus): The mechanism of injury in feline leukemia is virus-induced dysfunction and death and neoplastic transformation of lymphoid (hemopoietic) cells leading to (1) lymphoma (lymphosarcoma) and leukemia, (2) dysfunction of visceral organ systems, lymphoid tissues, or bone marrow, usually through compressive atrophy of parenchymal cells, in which the neoplastic cells proliferate, and (3) immunosuppression, resulting in increased susceptibility to other microbial diseases. Gross lesions include proliferation of neoplastic cells and their infiltration into perivascular spaces in organ systems resulting in (1) generalized enlargement of affected organs with increased pallor or (2) the formation of one or more solid white nodules distributed at random in the affected tissue (see Figs. 7-79, 13-79, and 13-85). Additionally, cells can occupy and proliferate in confined spaces, causing compressive atrophy of tissues such as axons in the spinal cord, hemopoietic cells in the bone marrow, and the retina in the eye.
Cats encounter virus in fomites from body fluids, such as salivary and nasal secretions, through direct contact with virus-infected cats. Virus is ingested or inhaled and is deposited on mucous members of the oral and nasal pharynx (preferentially the tonsils) and attaches to, infects, and replicates locally in mucosal epithelial cells and mucosa-associated lymphocytes and macrophages (MALT). It has not been determined how virus penetrates the mucus layer to gain access to mucosal epithelial cells or if mucosal macrophages and/or dendritic cells are involved in infection or spread locally. Virus spreads via leukocyte trafficking in lymphocytes and macrophages through lymphatic vessels to regional pharyngeal lymph nodes where it infects and replicates in additional lymphocytes and macrophages. B lymphocytes appear to be the primary cells used to spread virus via leukocyte trafficking, whereas T lymphocytes appear to be the primary target cell for infection and T lymphocyte dysfunction is involved in the symptomatology of the disease. From regional lymph nodes, virus spreads systemically in B lymphocytes via leukocyte trafficking to the circulatory system, through postcapillary venules or lymphatic vessels and the thoracic duct to lymph nodes and lymphoid organs, such as the spleen and Peyer’s patches, and then to bone marrow and mucosae of the salivary glands. Salivary gland secretions are an important mechanism to spread the virus. There are four subgroups of feline leukemia virus (FeLV) designated FeLV-A, FeLV-B, FeLV-C, and FeLV-T. FeLV-A is the subgroup transmitted between cats through saliva.
Once cats are infected with subgroup FeLV-A, the virus establishes a persistent infection (but not integrated in the genome) of bone marrow cells, which are most likely T lymphocytes or their precursor cells. Because viral replication occurs incessantly in these cells, there is greater opportunity for genomic variation to occur and new viral virulence determinates to be introduced. Subgroup FeLV-B appears to have arisen through recombination of endogenous genes of subgroup FeLV-A, whereas subgroup FeLV-C appears to have arisen through point mutations of endogenous genes of subgroup FeLV-A. Cats may be infected with only subgroup FeLV-A or a combination of subgroup FeLV-A with FeLV-B and/or FeLV-C. In general, subgroup FeLV-A causes immunosuppression and is found in approximately 100% of virus-infected cats; subgroup FeLV-B causes neoplastic transformation and is found in approximately 50% of virus-infected cats; and subgroup FeLV-C causes anemia and is found in approximately 1% to 2% of virus-infected cats. Recently, FeLV-T has been identified. It arises through genomic variation of FeLV-A, infects T lymphocytes, and causes an immunodeficiency syndrome.
Consideration of the encyclopedic listings of specific lesions and syndromes caused by feline leukemia virus is outside of the scope of this chapter; however, virus-induced lesions and syndromes include (1) lymphoma (lymphosarcoma) and all of its forms (alimentary, thymic, anterior mediastinal, multicentric, atypical) based on anatomic distribution, (2) leukemia, (3) myeloproliferative disorders, (4) nonregenerative anemia, (5) panleukopenia-like syndrome, and (6) glomerulonephritis. For successful replication, virus requires rapidly dividing cells such as lymphocytes. These syndromes arise from persistent infection of T lymphocytes in the bone marrow. Persistent infections result from modulation of virus and cellular gene expression and modification of the cat’s immune response by the virus. Persistence lasts for long periods, often the life of the cat, and occurs when the virus is not eliminated by the adaptive immune response because of dysfunction of cytotoxic T lymphocytes.
Immunosuppression and lymphopenia coincide with systemic involvement of lymphoid tissues, specifically T lymphocytes. Persistently infected cats commonly die of secondary bacterial and viral opportunistic infections. Subgroups A, B, C, and T use envelope surface glycoproteins to attach to receptors and enter T lymphocytes, other lymphocytes, and mucosal epithelial cells. Receptors for viral glycoproteins on these cells include (1) feline thiamine transport protein (FeTHTR1) as a receptor for FeLV-A; (2) feline phosphate transporter proteins 1 and/or 2 (FePit1 or FePit2) as receptors for FeLV-B; and (3) FeLV-C cellular receptor (FeLVCR), a heme transporter protein, as a receptor for FeLV-C. FeLV-T uses two proteins to attach to, enter, and infect cells. FePit1 is used as a receptor, whereas FeLIX, a protein secreted primarily by T lymphocytes, is used to restrict tropism to T lymphocytes. Retroviruses also have envelope glycoproteins that form multiple membrane-spanning glycoprotein systems that attach to and bind with multiple membrane-spanning receptors on lymphocytes. To infect T lymphocytes, FeLV-T expresses in its viral envelope a multiple membrane-spanning glycoprotein that attaches to and binds with a host cell multiple membrane-spanning receptor molecule (FePit1). It also appears that expression of a specific host cell receptor, the total number of receptors expressed on the cell, and the use of soluble cofactors play roles in determining which host cells are infected by FeLVs. Additionally, persistent infection of bone marrow cells by FeLV-A provides many opportunities for mutations in the envelope gene that result in the expression of new viral subgroups through viral envelope glycoproteins that recognize new host cell membrane receptors. It is likely that the clinical scenarios caused by these virus subgroups are related to genomic variation through the expression of envelope surface glycoproteins that determine and restrict which host cells they can attach to, enter, and infect.
Immunosuppression, targeted primarily to the cell-mediated immune system, appears to result from (1) a reduction in the number of lymphocytes, especially cytotoxic and helper cell T lymphocytes, through virus-induced cell death; (2) suppression of lymphokines (interferon-δ and interleukin secreted by activated T lymphocytes that could eliminate virus and virus-infected cells; (3) production of FeLV protein p15 that suppresses lymphocyte function (controversial); (4) dysfunction of lymphokine-induced activation of macrophages; and (5) dysfunction of neutrophil phagocytosis. Estimates suggest that about 50% of cats with certain bacterial infections and hemobartonellosis (Mycoplasma haemofelis) and 75% of cats with toxoplasmosis (Toxoplasma gondii) are infected with and suppressed by FeLV. Additionally, virus-induced immunosuppression has also been associated with feline infectious peritonitis, chronic oral and gingival diseases, poor reparative responses in inflammation, recurrent abscesses and skin infections, respiratory diseases, acute enteritides, otitis, and virus-induced malignancies such as sarcomas.
Neoplastic transformation follows persistent infection of T lymphocytes usually in bone marrow. Virus produces reverse transcriptase that transcribes viral RNA into proviral DNA and facilitates the insertion of proviral DNA into chromosomal DNA of T lymphocytes or other bone marrow cells. When virus has integrated its genome into the host cell DNA, viral genome is passed to all new generations of cells when the infected and integrated host cell is mitotic. Reverse transcriptase is carried by the virus and is released into host cell cytoplasm along with its viral RNA genome after the attachment and entry phase of the virus during its replication cycle. Neoplastic transformation of T lymphocytes or other bone marrow cells occurs when DNA provirus integrates into chromosomal DNA at critical regions that (1) contain oncogenes such as the cellular gene c-myc or (2) are near genes influencing the expression of c-myc genes. Activation of these genes and the expression of their gene products result in a series of alterations to the cell regulatory environment that leads to irreversible changes in cell behavior characteristic of neoplastic transformation (see Chapter 6). Feline oncornavirus-associated cell membrane antigen (FOCMA) is expressed on the cell membranes of transformed cells and is not found on normal (nontransformed) cells, even if they are infected with virus.
Feline Acquired Immunodeficiency Syndrome (Feline Immunodeficiency Virus, Lentivirus, Enveloped RNA Virus): The mechanism of injury in feline acquired immunodeficiency syndrome is provirus-induced dysfunction and death of CD4+ T lymphocytes leading to immunosuppression. Gross lesions include transient lymph node enlargement (lymphadenomegaly) followed by the occurrence of secondary opportunistic microbial infections. Feline immunodeficiency virus causes persistent and gradual depletion of CD4+ T lymphocytes (T helper (TH) lymphocytes, effector T lymphocytes, TH lymphocytes), resulting in an immunodeficiency syndrome characterized by chronic stomatitis and gingivitis, wasting syndrome (malnutrition), neurologic manifestations, and an increased incidence of lymphoma. The cause of CD4+ T lymphocyte depletion is unknown. It may have a multifactorial basis, including death of cells caused directly by viral infection, death (turnover) after massive and rapid replication of virus-infected and noninfected cells stimulated by viral antigen and/or inflammatory molecules, provirus-induced suppression of cell proliferation, death of provirus-infected CD4+ T lymphocyte by adaptive immune responses, or apoptosis of provirus-infected cells.
Cats encounter the virus in blood, most commonly as a provirus in infected CD4+ T lymphocytes, and much less commonly as free virus in fomites from saliva. During fights that result in bite wounds that bleed, blood contaminated with provirus-infected CD4+ T lymphocytes encounters (1) oral mucosae (macrophages and dendritic cells), especially of the tonsils through surface contamination and (2) macrophages and dendritic cells (Langerhans’ cells) of the skin through penetrating wounds. It appears that virus is able to establish a local infection in mucosal dendritic cells, macrophages, and lymphocytes; however, it is not clear how virus penetrates the mucus layer to gain access to mucosal epithelial cells, mucosal macrophages, and/or dendritic cells and migrates through the mucosal epithelium to reach cells in the submucosa (MALT). Hypothetically, several mechanisms of spread could be involved (1) migration (leukocyte trafficking) of provirus-infected CD4+ T lymphocytes through the epithelium into the submucosa, (2) infection of mucosal epithelial cells via the typical envelope-host cell receptor mechanism by virus released from provirus-infected CD4+ T lymphocytes, (3) infection of mucosal epithelial cells via the typical envelope-host cell receptor mechanism by cell-free virus, or (4) transfer of cell-free virus via viral transcytosis, the process by which virus is transported across the interior of a cell in vesicles to be released from the basal surface on the abluminal side. Blood in a skin wound would have direct access to Langerhans’ cells and tissue macrophages.
It appears that by whatever route used by virus to enter the body, it must gain access to local mucosal (MALT) or skin-associated lymphoid tissues (SALT) and CD4+ T lymphocytes, macrophages, and dendritic cells to establish an infection. Once these cells are infected, virus is then spread by leukocyte trafficking via lymphatic vessels to regional lymph nodes and then systemically via leukocyte trafficking to the spleen and other lymphoid tissues through postcapillary venules or lymphatic vessels and the thoracic duct. Some studies suggest that virus may also spread to the oral cavity and tonsillar mucosa via saliva either by provirus-infected CD4+ T lymphocytes or a cell-free viremia, especially if cats with chronic stomatitis and gingivitis are involved in grooming behavior or cat fights. Target cells for infection include CD4+ T lymphocytes, CD8+ T lymphocytes, B lymphocytes, cells of the monocyte-macrophage system, dendritic cells, megakaryocytes, and astrocytes. Virus envelope glycoproteins bind to host cell membrane receptors and serve to facilitate infection through virus attachment and entry into target cells. Little is known about the characteristics of envelope glycoproteins. Different stains of virus appear to express different envelope glycoproteins (and other proteins), thus these molecules likely contribute to viral pathogenicity. Target cells express feline CD134 receptor and CXCR4 cofactor in their membranes, both of which act as coreceptors and are needed for virus attachment, binding, and entry into host cells.
Postweaning Multisystemic Wasting Syndrome (Porcine Circovirus Type 2, Nonenveloped DNA Virus): The mechanism of injury in postweaning multisystemic wasting syndrome is virus-induced dysfunction and death of lymphocytes leading to lymphocyte depletion and immunosuppression. Virus appears to require dividing cells, like lymphocytes, in the S-phase of the cell cycle for infection and replication. Gross lesions include systemic enlargement of lymph nodes, normal size lymph nodes, and small atrophic lymph nodes, which are a continuum of changes in the response of lymphocytes to viral infection, replication, and release. Initial infection is likely correlated with viral replication and intense hyperplasia (lymphadenomegaly). Hyperplasia is followed by release of virus from infected lymphocytes, a process that kills lymphocytes and results in atrophy. Microscopic lesions are unique in the fact that inflammation is granulomatous with macrophage-derived syncytial giant cells.
Pigs encounter virus in fomites from oronasal-pharyngeal body fluids, feces, and urine from infected animals. Virus is inhaled or ingested and deposited on mucosae. In the respiratory system, virus is deposited on and trapped in the mucus layer by centrifugal and inertial turbulence and encounters mucosae of the tonsils. It has not been determined if and how virus penetrates the mucus layer to gain access to mucosal epithelial cells, mucosal macrophages, and/or dendritic cells. In the alimentary system, it is swallowed, gains access to the small intestine through peristalsis, and encounters M cell overlying Peyer’s patches. M cells lack a mucus layer, and virus has direct access to cell membranes.
It appears that virus establishes an infection in lymphoid tissues of the tonsil and Peyer’s patches by infecting mucosal dendritic cells, macrophages, and lymphocytes. Except for M cells, it is not clear how virus spreads through the mucosal epithelium to reach cells in submucosae (MALT), but leukocyte trafficking is likely involved. Spread through the mucosal epithelium could also occur through ligand-receptor interactions followed by viral transcytosis to the basal surfaces with release on the abluminal side. Once macrophages, dendritic cells, and lymphocytes are infected locally, virus spreads by leukocyte trafficking in macrophages and dendritic cells via lymphatic vessels to regional lymph nodes and then systemically through postcapillary venules or lymphatic vessels and the thoracic duct to the circulatory system to lymphocytes in the spleen, lymph nodes, and other lymphoid tissues.
Virus uses a viral capsid protein to attach to glycosaminoglycan receptors, heparin sulfate and chondroitin sulfate B, on macrophages, dendritic cells, and lymphocytes to enter and infect these cells. Macrophages are nonpermissive to virus and appear to serve primarily as trafficking cells to spread virus to other locations, whereas lymphocytes are permissive to virus and allow viral replication. Lymphocytes are injured and killed during replication. Although virus-induced necrosis has been suggested as the mechanism for cell death, apoptosis may actually be the main cause through a viral protein that activates caspase pathways. Other studies suggest that lymphoid loss may result from reduced production of lymphoid cells in the bone marrow or reduced proliferation in secondary lymphoid tissues, resulting in depletion of all types of T and B lymphocytes, immunosuppression, and increased susceptibility to secondary opportunistic infections. Although there is no proof that it is the causal agent, porcine circovirus type 2 (PCV2) has also been linked to several other conditions, including PCV2 pneumonia, PCV2 enteritis, PCV2 reproductive failure, and PCV2 porcine dermatitis and nephropathy syndrome. Many of these conditions have concurrent infections caused by other microbes. These conditions have been grouped under the term PCV2–associated diseases and will not be covered in this chapter because of limited information.
Nervous System
Canine Distemper (Morbillivirus, Enveloped RNA Virus): The mechanism of injury in canine distemper is dysfunction and death of epithelial, mesenchymal, neuroendocrine, and hematopoietic cells in many different tissues and organ systems. Gross lesions are not observed in the nervous system. In the bone marrow, blood cells, and lymphatic systems, they include lymphadenopathy followed by atrophy. Lymph nodes are initially enlarged, hemorrhagic, and edematous but are followed rapidly by cell death, resulting in loss of T and B lymphocytes in the spleen, lymph nodes, MALT, tonsil, and thymus. Alterations in bone marrow are minimal and nonspecific.
Dogs encounter virus in fomites from body fluids of the nasal and oral cavities, through direct contact with infected dogs. Virus is inhaled and deposited on mucosae of the conductive and O2-CO2 exchange systems through centrifugal and inertial turbulence. In the mucus layer of the oronasal pharynx, virus is phagocytosed by mucosal lymphocytes and macrophages and likely dendritic cells and spread via leukocyte trafficking to the tonsils. Here, lymphocytes and macrophages are infected and migrate in lymphatic vessels to regional lymph nodes, then systemically through postcapillary venules or lymphatic vessels and the thoracic duct to the circulatory system to lymphocytes in the spleen, thymus, lymph nodes, bone marrow, mucosa-associated lymphoid nodules and Peyer’s patches, and to Kupffer cells of the liver. Infection of cells may also occur via a cell-free viremia and through platelets. After infection of systemic lymphoid tissue, infected cells or virus spreads to parenchymal organs, including the nervous, respiratory, alimentary, and urinary systems. It infects a wide variety of epithelial and mesenchymal cells (pantropic virus) and kills these cells as it replicates and escapes from them.
In the respiratory system, virus kills pneumocytes, bronchiolar epithelium, and alveolar macrophages, thereby disrupting the function of the air-blood barrier, the mucociliary apparatus, and innate and adaptive immune responses resulting in poor oxygenation of blood, disrupted removal of particulate debris and secondary bacteria, and phagocytosis and antigen presentation by macrophages, respectively. These mechanisms contribute to suppurative bronchopneumonia. In the alimentary system, virus kills enterocytes (and likely M cells), leading to diarrhea. Virus also gains access to ameloblasts during the development of adult teeth, infects and kills these cells, and results in a condition known as enamel hypoplasia (see Fig. 7-17). Virus uses two viral envelope proteins: an attachment protein called viral H protein and a fusion protein called viral F protein bind to cell membrane glycoprotein receptors. Viral fusion proteins are involved in the penetration of virus into uninfected lymphocytes, spread of virus from cell-to-cell, and formation of syncytial cells (e.g., CD9 transmembrane protein) characteristically seen in the lungs. It has been shown experimentally that when virus-infected lymphocytes encounter uninfected lymphocytes and other cell types, they are induced to express new and/or increased numbers of SLAM receptors. Molecules secreted by virus-infected lymphocytes likely mediate this process and thus may serve as a means to amplify virus infection in dogs. Glycoprotein receptor CD150 (SLAM) occurs in membranes of lymphocytes, monocytes, macrophages, transitional epithelial cells, endothelial cells, and unspecified cells in the stomach, small intestine, and lung.
Vaccination status, adaptive immune system viability, and viral pathogenicity determine whether polioencephalomyelitis, demyelinating leukoencephalomyelitis, or both occur in the CNS. Virus-infected lymphocytes and macrophages spread distemper virus via the blood vascular system to the CNS through leukocyte trafficking and cell-free viremia. Cells and virus likely interact via the leukocyte adhesion cascade (see Chapter 3) with endothelial cells by adhering to and migrating through the endothelium. Virus also infects and replicates in endothelial cells of capillaries and postcapillary venules, resulting in a perivascular lymphomonocytic inflammatory response characteristic of viral infections. Virus then infects and replicates in vascular pericytes, microglial cells, and perivascular astrocytic foot processes, as well as in choroid plexus epithelium. Virus escapes from choroid plexus epithelium and spreads in the cerebral spinal fluid (CSF) to infect ependymal cells and oligodendroglial cells in the subependymal white matter. The clinical signs caused by virus are likely related to injury of neurons and oligodendroglial cells. The virus can cause disease in both gray matter (neurons: polioencephalomyelitis) and white matter (oligodendroglial cells: demyelinating leukoencephalomyelitis). Neuronal infection likely arises after spread of virus to neurons from virus-infected pericytes and perivascular astrocytic foot processes. Virus-infected astrocytes may also serve as a reservoir for spreading virus within the CNS. Viral infection of neurons results in neuronal necrosis and subsequent neuronophagia via resident microglial cells and trafficking monocytes, macrophages, and lymphocytes. Spread of virus to oligodendroglial cells probably arises from infection of ependymal cells; however, infection through the blood vascular system, capillaries and postcapillary venules, and virus-infected pericytes and perivascular astrocytic foot processes has not been excluded as a potential infective mechanism.
Involvement of oligodendroglial cells results in demyelinating leukoencephalomyelitis, which has an acute phase and a chronic phase. Two mechanisms have been proposed for the acute phase of demyelinating leukoencephalomyelitis: (1) death of oligodendroglial cells from infection or (2) a type II hypersensitivity reaction against proteins such as myelin basic protein and myelin-associated glycoprotein. For a cell death mechanism, there is no evidence of virus-induced apoptosis or necrosis of oligodendroglial cells, and although virus can infect oligodendroglial cells, no viral proteins are present in these cells. Astrocytes and microglial cells can be infected and show activation such as hypertrophy and hyperplasia. It has been hypothesized that toxic molecules, such as proinflammatory cytokines produced by these glial cells, act to disrupt the function of oligodendroglial cells and kill the cells. For a hypersensitivity reaction mechanism, microscopic lesions of vacuolization (intramyelinic edema) of myelin lamellae surrounding axons in white matter accompanied by reactive astrocytes, macrophages (monocytes), resident microglial cells, and occasional multinucleated giant cells are consistent with this type of immune-mediated injury. As this injury progresses, the inflammatory response becomes more intense and is characterized by perivascular mononuclear infiltrations. Myelin is phagocytosed by macrophages (monocytes) and microglial cells and the lesion is repaired by proliferation of astrocytic processes, thus forming dense plaques (astrocytic scars). The chronic phase of demyelinating leukoencephalomyelitis appears to be a bystander mechanism involving inflammation and virus-induced immune responses, such as antibody dependent cell-mediated reactions (cytotoxic T lymphocytes) against viral proteins expressed in oligodendroglial cell membranes leading to macrophage-mediated separation, damage, and phagocytosis of myelin lamellae. Myelin damage is likely the result of proteolytic enzymes, oxygen free radicals, and cytokines from activated macrophages, monocytes, and resident microglia. Lipids from damaged lamellae stimulate an intense phagocytic response and likely initiate recruitment of additional monocytes and macrophages into the lesions. Disruption of the blood-brain barrier by proteolytic enzymes appears to play a role in the influx of inflammatory cells probably mediated by viral infection of astrocytes through their foot process involved in the structure and function of the blood-brain barrier.
Rabies (Lyssavirus, Enveloped RNA Virus): The mechanism of injury in rabies is neuronal dysfunction likely caused by several proposed mechanisms such as viral takeover of RNA transcription and translation in neurons, disruption of neurotransmitter functions, dysfunction of ion channels, and/or induction of the synthesis of nitric oxide. Rabies virus infects neurons of all mammalian species. Gross lesions are not present in nervous tissue; however, inclusion bodies (Negri bodies) and a chronic lymphomonocytic perivascular inflammation characteristic of viral infections are observed (see Figs. 1-49,C and 14-45). In addition to neurons, the virus infects glial cells in the nervous system and epithelial cells such as those in the salivary glands.
Animals encounter virus in fomites from saliva through a skin-penetrating bite wound from a rabid animal. Virus gains access to interstitial (extracellular) body fluids and plasma (bite wound hemorrhage); diffuses at random in this fluid; and encounters, attaches to, and enters striated muscles cells via binding of rabies virus envelope G protein to neurotransmitter receptors, such as an acetylcholine receptors, in cell membranes. Envelope G protein is an important determinate of rabies neurovirulence and which neuron pathways are infected with virus in the nervous system. Virus replicates in muscle, buds from cell membranes, enters interstitial fluids of myoneural junctions, and randomly encounters and binds to acetylcholine receptors, neuronal cell-adhesion molecule receptors, neurotrophin receptors, or other types of gangliosides in cell membranes of unmyelinated axon terminals (nerve endings) of lower motor neurons or sensory neurons of peripheral nerves. Similar processes are also used to spread and replicate virus in cranial nerves after bite wounds to the face. Once bound, virus enters the cytoplasm of nerve endings through pinocytosis via clathrin-coated pits and the formation of vesicles. Virus in vesicles spreads centripetally from myoneural junctions to the cell body of the nerve via retrograde fast axonal transport, likely using the dynein light chain microtubule-based transport system. Virus replicates in the cell body of neurons and travels to dendrites via axonal transport where it buds from cell membrane of dendritic processes into synaptic clefts of neural-neural junctions. It randomly encounters receptors on sensory and motor nerve endings within the spinal cord and brain in gray horns of the spinal cord. Mechanistically, viral replication and spread in sensory and motor neurons within the spinal cord and brain are identical to that in peripheral spinal nerves. The exact mechanism that facilitates transsynaptic spread of rabies virus is unknown. It may be linked in part to viral assembly where M-protein encapsulates the virus and assists in moving the virus to cell membranes such as those in synapses that contain glycoproteins essential for formation of the viral envelope and viral budding. Envelope G protein is also required for attachment to cell membrane and transsynaptic spread of the virus to the next neuron in the neural pathway.
Virus uses axonal transport mechanisms to spread throughout the body via afferent and efferent neural pathways to infect the epithelial cells of the salivary glands (see Fig. 14-44). Rabies virus, through these neural pathways, can also infect other cells such as those in taste buds, nasal cavity, skin and hair follicles, adrenal gland, pancreas, kidney, heart muscle, and the retina and cornea. In fact, the “furious” and “dumb” forms of rabies in domestic animals are likely attributable to infection of specific neuronal populations and pathways such as those in the hippocampal formation or cerebellum, respectively. Virus spreads to salivary glands through axonal transport using parasympathetic nerves present in the facial (VII) and glossopharyngeal (IX) cranial nerves and sympathetic nerves in the thoracic segments of T1-T3 spinal cord segments. In addition to spreading virus to the salivary glands, viral infection of parasympathetic and sympathetic nerves also results in increased salivary gland secretions: (1) directly through stimulating β-adrenergic receptors on the salivary acinar and ductal cells, leading to an increase in cAMP concentrations and the corresponding increase of saliva secretion, and (2) indirectly through stimulating nerves innervating blood vessels that supply the salivary glands. Virus buds from the cell membrane of these nerve terminals, infects salivary acinar cells through the envelop G protein-specific cell surface receptor mechanism, and replicates in and is amplified to large quantities in salivary acinar cells. Virus then buds from apical (luminal) surfaces of acinar cell membranes, mixes with saliva, and can be transmitted in a bite wound. The apical specificity of viral budding is established during the assembly stage of viral replication. Viral genome and proteins form complexes in the acinar cell cytoplasm that congregate at areas of the cell membrane that contain matching glycoprotein receptors and then bud from this membrane into the acinar lumen.
Equine Polioencephalitis-Polioencephalomyelitis (Alphavirus, Enveloped RNA Virus): The mechanism of injury in equine polioencephalitis-polioencephalomyelitis is disruption and death of neurons in the CNS. Gross lesions include active hyperemia, vasculitis, hemorrhage, and yellow-white-gray areas of necrosis in gray matter of the nervous system, especially the spinal cord (see Fig. 14-79). Because neurons are the primary target, lesions are most commonly observed in gray matter, areas in which neuron cell bodies are located, and these diseases are classified as polioencephalitides or polioencephalomyelitides. Equine polioencephalitis-polioencephalomyelitis is used to group three closely related strains of alphaviruses that cause Eastern equine encephalomyelitis, Western equine encephalomyelitis, and Venezuelan equine encephalomyelitis. St. Louis encephalomyelitis is the human counterpart to these horse diseases. Such diseases have also been called arbovirus polioencephalitis-polioencephalomyelitis. The term arbovirus is derived from the fact that these viruses are arthropod-borne; the term was shortened and is used as a disease acronym.
Horses encounter viruses through skin penetrating bite wounds from virus-infected mosquitoes. Mosquitoes are infected by biting birds, the reservoir for the virus. Seasonal variations in temperature and precipitation greatly influence the population density of mosquitoes and thus the occurrence of disease. After skin penetration, virus can either directly enter the circulatory system and infect monocytes or be deposited in vascularized ECM (connective) tissue and infect dendritic cells (Langerhans’ cells) and tissue macrophages (Fig. 4-45). In these cells, virus is spread via leukocyte trafficking to regional lymph nodes either by the circulatory system or lymphatic vessels where it infected lymphocytes. It may also spread to regional lymph nodes via cell-free viremia in lymphatic vessels. Viral envelope contains two membrane-anchored glycoproteins, E1 and E2. Attachment protein E2 is used to attach to target cell receptor, whereas viral envelope fusion protein E1 is used to enter cells via endocytosis. Receptors for E1 and E2 proteins occur on a variety of cell types and probably determine which organ systems, such as the nervous system, are targeted for infection by virus. Virus then spreads systemically via leukocyte trafficking in lymphocytes and macrophages through postcapillary venules or lymphatic vessels and the thoracic duct to the circulatory system to systemic lymph nodes, spleen, thymus, bone marrow, Peyer’s patches, pancreas, and skeletal muscle. Infection results in necrosis of myeloid cells in bone marrow and lymphocytes in lymph nodes and spleen. Proinflammatory cytokines, such as IFN-γ, and antiinflammatory cytokines, such IL-10 produced by infected lymphocytes, may cause cell death. Cytokines released into the blood vascular system may also act on the blood-brain barrier, making it more susceptible to infection. In Eastern equine encephalomyelitis, osteoblasts appear to be the systemic population of target cells used to amplify virus so it can spread to the nervous system, whereas dendritic cells, lymphoid cells, and cells of the monocyte-macrophage system are not as susceptible to infection and thus systemic lymph nodes and spleen are infected to a limited degree with minimal injury. Although it is unclear how virus spreads to and enters the CNS, leukocyte trafficking by lymphocytes and macrophages (monocytes) appears to be the probable mechanism. Cell-free viremia may also occur.
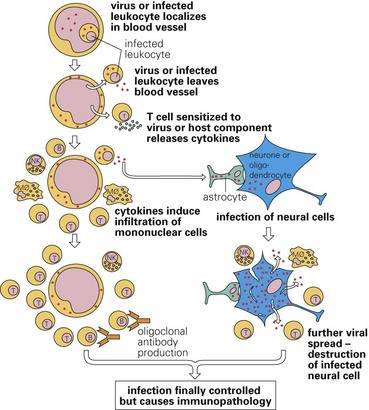
West Nile Virus Polioencephalitis-Polioencephalomyelitis (Flavivirus, Enveloped RNA Virus): The pathogenesis and mechanism of injury in West Nile virus polioencephalitis-polioencephalomyelitis are similar to those of equine polioencephalitis-polioencephalomyelitis (see Fig. 4-45).
Bovine Herpesvirus Meningoencephalitis (Bovine Herpesvirus 5: Alphaherpesvirus, Enveloped DNA Virus): Bovine herpesvirus 5 infects cells, spreads, and behaves much like bovine herpesvirus 1, except it is more neurovirulent and induces a severe often fatal encephalitis. See infectious bovine rhinotracheitis in the section on the Respiratory System, Mediastinum, and Pleurae. The mechanism of injury is dysfunction and death of neurons and astrocytes caused by viral replication and likely the actions of cytotoxic T lymphocytes on virus-infected neural cells through proinflammatory chemokines and cytokines as part of lymphomonocytic inflammation (innate and adaptive immune responses). Gross lesions include randomly distributed cerebral edema, active hyperemia, hemorrhage, and malacia.
Cattle encounter virus in fomites from body fluids through direct contact with virus-infected animals. It is inhaled or ingested and deposited on mucosae of the oral, nasal, and pharyngeal cavities and of the conjunctiva or inhaled and deposited on mucosae of the conductive component of the respiratory system through centrifugal and inertial turbulence. Viral envelope glycoproteins B, C, D, and E are used to attach to receptors on sensory nerve endings that innervate these mucosae. They can also attach to receptors on a variety of other target cells. These receptors are glycosaminoglycan receptors such as herpesvirus entry mediator A, nectin-1 and -2 (herpesvirus entry proteins C and B), and 3-O-sulfated heparin sulfate. It has not been determined how virus penetrates the mucus layer to gain access to mucosal sensory nerve endings. Through these nerve endings, virus enters neurons, such as the trigeminal and olfactory nerves, and spreads via retrograde axonal transport to other neurons and glial cells within the nervous system. It appears that envelope glycoprotein E and 3-O-sulfated heparin sulfate receptors may amplify viral attachment, entry, and spread within the CNS. The mechanism of malacia remains unknown but apparently is not caused by obvious vascular injury. Neuronal lesions are consistent with necrosis, likely caused by virus-induced injury and cell death. However, overproduction of nitric oxide in virus-infected neurons and astrocytes could result in their dysfunction and death and that of contiguous non-infected cells.
Bovine herpesvirus 5 can enter latency in the nervous system, through mechanisms likely identical for bovine herpesvirus 1.
Equine Herpesvirus Myeloencephalopathy (Equine Herpesvirus 1: Alphaherpesvirus, Enveloped DNA Virus): The mechanism of injury in equine herpesvirus myeloencephalopathy is dysfunction and death of endothelial cells in small arterioles of the brain and spinal cord; however, the means is uncertain but most likely caused by virus replication. Immune-complexes (type III hypersensitivity reaction) and the fixation of complement (immune complex-induced vasculitis) have also been suggested. Gross lesions include randomly distributed foci of edema, hemorrhage, and vasocentric malacia (yellow-white-gray areas) consistent with vascular occlusion, resulting in infarction (see Fig. 14-80).
Horses encounter virus in fomites from body fluids through direct contact with virus-infected animals. It is inhaled or ingested and deposited on mucosae of the oral, nasal, and pharyngeal cavities or inhaled and deposited on mucosae of the conductive component of the respiratory system through centrifugal and inertial turbulence. Virus infects and replicates in mucosal epithelial and endothelial cells, then in contiguous mucosal and submucosal lymphocytes and likely macrophages, monocytes, and dendritic cells (MALT), and then spreads via leukocyte trafficking in lymphatic vessels to regional lymph nodes. It has not been determined if and how virus penetrates the mucus layer to gain access to mucosal epithelial and endothelial cells or if or how mucosal macrophages and/or dendritic cells are involved, although very likely. Although specific ligands and receptors have not been identified, viral envelope glycoproteins likely attach to glycosaminoglycan receptors on host cell membranes and use this binding to enter the cells listed above. Infection appears to be sustained and amplified in lymphocytes and likely macrophages and monocytes of regional lymph nodes and then is spread systemically through blood and lymphatic vessels in these cells via leukocyte trafficking into the circulatory system. Infected cells probably use envelope adhesion molecules to bind to receptors on vascular endothelium and during migration through the vessel wall, interact with these cells, allowing virus to infect and replicate in endothelial cells, myocytes, and pericytes of small arterioles in the brain and spinal cord causing vasculitis and thrombosis. It is not known why these cells are targets for virus infection; however, typical ligand-receptor interactions or permissiveness of these cells are likely involved. Activation of endothelial and leukocyte adhesion molecules is an important step in spreading virus to endothelial cells and thus may contribute to endothelial cell tropism for viral infection.
Parvovirus-Induced Cerebellar Hypoplasia (Parvovirus, Nonenveloped DNA Virus): See parvovirus enteritis in the section on the Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity for information about local and regional stages of viral spread and replication before spread to the CNS. In pregnant cats, parvovirus is able to cross the placenta and infect dividing cells in the developing cerebellum in kittens, resulting in cerebellar hypoplasia (see Fig. 14-35). Whether by leukocyte trafficking or cell-free viremia, parvoviruses are able to gain access to cells in the placenta. Virus infects and replicates in placental trophoblasts and spreads to, infects, and replicates in cytotrophoblasts and cells of the mesenchymal stroma of the fetal placenta. From these cells, virus gains access to the fetal vascular system and spreads to, infects, and replicates in hematopoietic cells and other dividing cells. It has also been suggested that placental macrophages and fetal endothelial cells are likely involved in the replication and spread of the virus in the fetus. Although the virus can infect a large number of different cells in the fetus, it is unclear why fetal infection is clinically dominated by injury to cells of the cerebellum and cerebellar hypoplasia. Ligand-receptor interactions could contribute to specificity; however, other unknown mechanisms are likely involved.
Parvoviruses only infect and replicate in dividing cells. Cells of the external granular layer of the cerebellum are dividing cells, whereas Purkinje cells are nondividing cells. However, cell death is observed in both of these cell types, when only one of them is a dividing cell population. Granule precursor cells of the cerebellar external granular layer are the major target cells for parvovirus replication during the perinatal period because they are able enter the S-phase of the mitotic cycle. Purkinje cells are also infected, but they are nondividing postmitotic cells. It appears that virus infects Purkinje cells via a host cell membrane transferrin receptor that is commonly used by parvovirus for entering other types of host cells. Virus is unable to replicate in postmitotic Purkinje cells, but transcription of viral proteins does occur. It has been suggested that a nonstructural parvovirus protein NS1 is produced at low concentrations during the G0 and G1 phases of the cell cycle. Because NS1 is known to be highly cytotoxic and able to induce cytoskeleton alterations, this effect could result in injury and cytolysis of Purkinje cells during in utero infection with virus. Although cerebellar hypoplasia is not commonly thought to occur from in utero infection of female dogs by canine parvovirus, a recent study has identified parvovirus DNA in brain tissue from puppies with the disease. However, the significance of this information remains unclear because parvovirus structural proteins were not identified in the same tissues. A similar syndrome, probably involving similar mechanisms, occurs in calves infected in utero with bovine viral diarrhea–mucosal disease virus (see Fig. 14-36).
Visna (Maedi-Visna Virus [Ovine Lentivirus], Enveloped RNA Virus): The chronologic sequence of events that characterizes the pathogenesis of injury in visna is similar to those that occur in progressive ovine pneumonia (Maedi) of sheep. The mechanism of injury is chronic-active (granulomatous) inflammation of the CNS resulting in demyelinating encephalitis. Gross lesions include foci of yellow-white malacia distributed at random in the CNS. Ovine lentivirus persistently infects cells of the monocyte-macrophage system including microglial cells (local tissue macrophages in the CNS) and all of these cell types are central to the genesis of the inflammatory response in the CNS.
Caprine Encephalitis (Caprine Arthritis-Encephalitis Virus, Enveloped RNA Virus): The pathogenesis and mechanism of injury in caprine encephalitis are similar to those that occur in progressive ovine pneumonia (Maedi) of sheep (see section on the Respiratory System, Mediastinum, and Pleurae); however, the initial route of exposure is different. The mechanism of injury is chronic-active (granulomatous) inflammation of the CNS resulting in demyelinating myelitis. Gross lesions include foci of yellow-white malacia distributed at random in the CNS, especially the spinal cord (see Fig. 14-90). Caprine arthritis-encephalitis virus persistently infects cells of the monocyte-macrophage system, thus microglial cells (local tissue macrophages) and trafficking monocytes serve as the cell type central to the genesis of the inflammatory response. Kid goats are primarily exposed to virus through the ingestion of virus-infected milk or colostrum. Although not proven, virus likely infects M cells overlying Peyer’s patches. Once infected, virus is transferred to and released from basilar surfaces of M cells to gain access to macrophages and lymphocytes within Peyer’s patches. It is here that macrophages are infected with virus and then serve to spread virus to monocyte precursor cells in the bone marrow and ultimately to the CNS.
Pseudorabies (Aujeszky Disease) (Alphaherpesviruses, Enveloped DNA Virus): The mechanism of injury in pseudorabies is disruption and death of neurons likely caused by the actions of cytolytic immune cells interacting with virus-infected neurons. Because neurons are the primary target of viral infection, lesions are most commonly observed in gray matter and as a result, this disease is a polioencephalitis or polioencephalomyelitis. Gross lesions characteristic of injury are usually not observed but in severe cases could include active hyperemia and hemorrhage.
Pigs encounter virus in fomites from oronasal-pharyngeal body fluids most commonly through inhalation and potentially through contamination of skin-penetrating bite wounds. When inhaled, virus is deposited on mucosae of the oral, nasal, and pharyngeal cavities, especially of the tonsil or on mucosae of the conductive component of the respiratory system through centrifugal and inertial turbulence. In the tonsil, virus may infect and replicate in mucosal epithelial cells, mucosal and submucosal macrophages, and dendritic cells (MALT). In the lung, virus also infects and replicates in similar cells (BALT), including alveolar macrophages, which it kills, resulting in a secondary bronchopneumonia. Virus attachment and entry is likely mediated by binding of viral envelope glycoproteins to host cell membrane receptors. In the nasal and pharyngeal mucosae and submucosae, especially of the tonsil, virus encounters and infects sensory nerve endings of the olfactory, glossopharyngeal, and trigeminal cranial nerves and uses retrograde axonal transport to enter the brain. Virus can spread transsynaptically throughout the CNS by using mechanisms similar to those described in rabies and infect and replicate in many types of neurons. Viral envelope glycoproteins C, B, D, H, and L are used to attach to, fuse with, and enter membranes of nerve endings. These glycoproteins are also involved in transsynaptic spread to other neurons in the CNS and to other neural cells, such as astrocytes, microglial cells, ependymal cells, and trafficking monocytes/macrophages, as well as in the formation of syncytial cells and the modulation of innate and adaptive immune responses. Virus cannot replicate in these cells, thus they are incapable of infecting other cells in the CNS. This outcome may represent a local intrinsic and/or innate immune defense mechanism that isolates through phagocytosis the virus in astrocytes, monocytes-macrophages, and microglial cells and restricts spread of virus to other cells. Latent infections involve the trigeminal nerves and ganglia, but tonsillar lymph nodules may also be involved. Potentially, peripheral nerve endings in the skin, subcutis, and muscle may be exposed to infection via bite wounds and can be used by virus to gain access to and enter the CNS by mechanisms similar to those described in rabies.
Viral envelope glycoproteins in membranes of infected neurons are targets for neutralizing antibodies, cytotoxic T lymphocytes, and lymphokine-activated killer cells and are part of the chronic perivascular lymphomonocytic inflammatory response characteristic of viral infections. These cells may contribute in a large manner to neuronal injury and death in pseudorabies. Hypertrophy and hyperplasia of astrocytes, microglia, and monocytes-macrophages occur spatially and temporally with the severity of neuronal injury; however, the potential role of biologically active molecules, such as cytokines (e.g., TNF-α), from these cells is unclear.
Bone, Joints, Ligaments, and Tendons
Caprine Arthritis (Caprine Arthritis-Encephalitis Syndrome, Enveloped RNA Virus): The mechanism of injury in caprine arthritis is chronic-active (granulomatous) inflammation of the synovium, resulting in proliferative synovitis. The chronologic sequence of events that characterizes the pathogenesis of injury in caprine arthritis is similar to those that occur in progressive ovine pneumonia (Maedi) of sheep (see section on the Respiratory System, Mediastinum, and Pleurae).
Integumentary System
Pox (Cowpox [Orthopoxvirus], Sheeppox and Goatpox [Capripoxvirus], Swinepox [Suipoxvirus], Enveloped DNA Virus): The term pox is used to group diseases, such as bovine cowpox, sheeppox, goatpox, swinepox, and lumpy skin disease, that are caused by closely related strains of poxviruses. The mechanism of injury is dysfunction and death of dendritic and epithelial cells of the skin. Gross lesions include macules, papules, vesicles, pustules, scabs, and scars (see Figs. 17-31, 17-42, and 17-44). Lesions are most easily observed on wool-free or hair-free areas (Fig. 4-46). In general, sheeppox and goatpox are more virulent and cause systemic disease, whereas bovine cowpox and swinepox usually do not cause systemic disease. In these latter species, spread of virus is the result of animal-to-animal contact or contact with clothing or tools/instruments contaminated with virus-infected skin, scabs, or other skin debris. It appears that skin must be injured (traumatic abrasions) so that capillary endothelial cells, trafficking leukocytes, or Langerhans’ cells (dendritic cells) are exposed and can encounter virus.
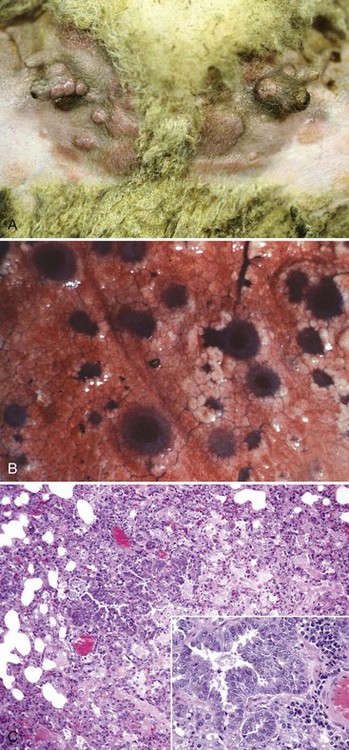
Fig. 4-46 Sheeppox and goatpox.
A, Skin, teats, inguinal area. Macules, papules, vesicles, crusts (scabs), and papillomas (epidermal hyperplasia) are present on the skin of the inguinal area and teats. Additional information about the development and progression of pox virus-induced lesions is schematically illustrated in Fig. 17-31 and macroscopically and microscopically in Figs. 17-42 (sheeppox) and 17-44 (swinepox). B, Lung, pox lesions. These circumferentially expanding dark red to plum-colored lesions of varied sizes are areas of proliferating bronchial and bronchiolar mucosal epithelial cells, necrotic epithelial cells, cell debris, and inflammation demonstrated in C. C, Lung, bronchiole. There is proliferation of mucosal epithelial cells of the lung’s conductive system that are infected with poxvirus. Note the mononuclear inflammatory likely bronchiole-associated lymphoid tissue (BALT) in adjacent supporting stroma. Inset, Higher magnification of C. H&E stain. (A courtesy Dr. D. Gregg, Plum Island Animal Disease Center and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. R. Breeze, Plum Island Animal Disease Center and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. C courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
As examples, bovine cowpox most commonly occurs on the teats of dairy cows, the areas most commonly injured by milking trauma in a diary herd. Insect bites result in penetrating skin wounds that can also carry virus into contact with susceptible target cells. However, in sheeppox and goatpox, animals encounter virus through the oronasal pharynx via inhalation or ingestion. It is deposited on mucosae, especially of the tonsil, and infects and replicates in epithelial cells, mucosal lymphocytes and macrophages, and dendritic cells (MALT). It has not been determined how virus penetrates the mucus layer to gain access to mucosal epithelial cells, macrophages, and/or dendritic cells, but it is likely virus is phagocytosed by leukocytes trafficking in the mucus layer when these cells encounter virus during migration. Submucosal macrophages are infected, and virus spreads in them via lymphatic vessels to regional lymph nodes, such as the submandibular and pharyngeal. Here, naïve lymphocytes and macrophages are recruited through the release of proinflammatory chemokines and cytokines and are infected with virus. Virus then spreads systemically via leukocyte trafficking in these lymphocytes and macrophages through postcapillary venules or lymphatic vessels and the thoracic duct to the circulatory system and then to systemic lymph nodes, spleen, and bone marrow and infects and replicates in similar cells. Virus then spreads from systemic lymphoid tissues via leukocyte trafficking to the skin (see below), lung, liver, and other organ systems.
In the skin, virus spreads from migrating macrophages and lymphocytes and infects and replicates in endothelial cells, resulting in direct injury and an acute inflammatory response. Endothelial cell injury accompanied by vascular dilation, active hyperemia, and acute inflammation in part likely account for macules and papules observed in early skin lesions. Langerhans’ cells (dendritic cells) are in close contact with endothelial cells in the Malpighian layer of the skin. It appears that virus from capillary endothelial cells and trafficking leukocytes is able to infect Langerhans’ cells and then spread virus to contiguous skin epithelial cells of the stratum basale and spinosum. All of these cells allow virus to replicate, thus when epithelial cells of the stratum basale and spinosum are killed, the space formerly occupied by these cells coalesces and is filled with cell debris and intercellular edema, forming vesicles. With injury, acute inflammation ensues, as does the pustular stage. Through adaptive immune responses, viral infection is resolved and pustular lesions heal as scabs over granulation tissue that become scars.
It is likely that both humoral and cell-mediated immunity are important in protecting against and resolving pox diseases; however, these responses can cause injury and death of virus-infected host cells. Similar lesions and lesion progression may affect oral mucous membranes. Pneumonia has been reported in systemic poxvirus-induced disease. Affected lungs have variable sized and randomly distributed pock lesions in the form of large, irregularly shaped lobular areas of consolidation (see Fig. 4-46). This pattern is consistent with hematogenous spread of the virus via leukocyte trafficking in virus-infected macrophages to pulmonary endothelial cells and then to bronchiolar and alveolar epithelial cells followed by cell death and acute inflammation. Although reservoir hosts for poxvirus are wild rodents, cats are now the most commonly recognized reservoir. Cats are infected with virus through their skin by an indirect mechanism when hunting virus-infected rodents; however, infection, as previously described, via a direct mechanism (inhalation) and systemic spread in monocytes and macrophages has been reported.
Poxviruses use attachment proteins to bind to glycosaminoglycan receptor proteins on the surface of the host target cells. Because of the volume of information related to attachment proteins and receptors in poxvirus diseases, discussion of these protein molecules is outside of the scope of this chapter.
Contagious Ecthyma (Orf Virus: Parapoxvirus, Enveloped DNA Virus): See contagious ecthyma in the section on the Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity.
Bovine Papular Stomatitis (Parapoxvirus, Enveloped DNA Virus): See bovine papular stomatitis in the section on the Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity.
Vesicular Stomatitis (Vesiculovirus, Enveloped RNA Virus): See vesicular stomatitis in the section on the Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity.
Swine Vesicular Disease (Enterovirus, Nonenveloped RNA Virus): See swine vesicular disease in the section covering the Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity.
Vesicular Exanthema of Pigs (Calicivirus [Waikavirus], Nonenveloped RNA Virus): See vesicular exanthema of pigs in the section on the Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity.
Foot-and-Mouth Disease (Aphthovirus, Nonenveloped RNA Virus): See foot-and-mouth disease in the section on the Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity.
Viral Papillomas (Warts, Sarcoids, Papillomaviruses, Nonenveloped DNA Virus): The mechanism of injury in viral papillomas is dysfunction of host cell genes that regulate cell proliferation, differentiation, and adhesion, resulting in benign neoplastic transformation of virus-infected epithelial cells. Cells of stratum basale (germinativum) play a central role in the pathogenesis of neoplastic transformation. Gross lesions include the formation of exophytic and occasionally endophytic papillomatous fronds that arise from mucosae or skin (see Fig. 17-45). Papillomaviruses are species specific and in (1) cattle cause warts of the skin and papillomas of mucosae of the alimentary system, teats and udder, and penis; (2) horses, donkeys, and mules cause sarcoids of the skin; and (3) dogs cause papillomas of the mucosal epithelium of the oral cavity and reproductive system.
Animals encounter virus through direct contact with animals of the same species having warts, papillomas, or sarcoids. Viral infection is preceded by injury of the superficial layers of the stratified epithelium of mucosae or skin, resulting in exposure of target cells in the stratum basale. Epithelial cells of mucosae have short lifespans and with aging are shed from their surfaces into a lumen of the alimentary system or into the environment as a means of spreading the virus. Thus these cells are constantly being turned over and replenished by dividing stem cells located in their basal layers. Maturation of these cells begins with the least differentiated layer, the stratum basale, and progresses outwardly through the suprabasilar layers, the strata spinosum, granulosum, lucidum, and corneum. Stem cells of the stratum basale are continuously dividing to replace cells in the suprabasilar layers; cells of the suprabasilar layers do not divide. Virus likely uses capsid proteins, such as bovine L1 major capsid protein and L2 minor capsid protein, to attach and bind to and enter cells of the stratum basale. Viral receptors on cells of the stratum basale have not been clearly identified; however, heparin sulfate proteoglycans mediate the initial attachment of virions to cells in some experimental systems. Dividing cells of the stratum basale are target cells for viral infection and it replicates its genome to a limited extent within the nucleus of these cells. Stem cells of the stratum basale have a lifespan for the life of the animal and serve as reservoirs for virus-infected cells. They also are nonpermissive cells, and virus is not able to produce infective virions. As cells of the stratum basale mature through differentiation into cells of the strata spinosum, granulosum, lucidum, and corneum, these differentiated cells become permissive and allow virus to complete its replication cycle and produce infective virions. Virus is released from cells of the strata lucidum and corneum into the environment to spread the disease. A similar process likely occurs in infected mucosae of the alimentary system.
Neoplastic transformation of epithelial cells by papillomaviruses can result in the formation of benign tumors, such as papillomas, warts, and sarcoids, and malignant tumors, such as carcinomas. When virus infects stems cells of the stratum basale, the expression of viral genes is maintained at low numbers (approximately 20 to 100 extrachromosomal copies of viral DNA per cell) where it replicates in synchrony with the cell cycle as the cell divides. Normally, as epithelial cells leave the stratum basale and mature (differentiate), they turn off endogenous genes and the synthesis of proteins required for cell division. When virus-infected stem cells of the stratum basale divide, viral genomes are carried in cells that differentiate into cells of the suprabasilar layers. Viral proteins prevent these differentiated cells from stopping the cell cycle, thus cells of suprabasilar layers, especially the strata spinosum and granulosum, are now capable of division. Because cells of the suprabasilar layer are permissive and allow virus to complete its replication cycle and produce infective virions, large quantities of viral genes and regulatory proteins are present within these dividing host cells. As a general rule, neoplastic transformation of virus-infected epithelial cells appears to be linked to the quantitative and qualitative expression of viral genes and gene products, such as oncoproteins, and how these molecules interact with host cell genes and gene products regulating cell proliferation, differentiation, and adhesion. It looks as if strains of papillomavirus unable to integrate into host genes are most likely to cause benign transformation (papillomas, warts, and sarcoids) of virus-infected epithelial cells, whereas strains able to integrate into host genes are most likely to cause malignant transformation (carcinomas) of virus-infected epithelial cells. Benign transformation is most likely to occur in cells of the stratum basale. In these cells, virus does not integrate into host cell genes and viral genes and gene products like oncoproteins are expressed in low numbers. Thus the likelihood of virus activating growth-promoting genes (oncogenes) in host cell DNA, inactivating suppressor genes that would inhibit cell proliferation, and altering the functional expression of genes that regulate apoptosis is extremely limited. Malignant transformation is most likely to occur in suprabasilar cells where virus integrates into host cell genes and viral genes and gene products, such as oncoproteins, are expressed in high numbers. Thus the likelihood of virus activating growth-promoting genes (oncogenes) in host cell DNA, inactivating suppressor genes that would inhibit cell proliferation, and altering the functional expression of genes that regulate apoptosis is extremely high. A similar process likely occurs in infected mucosae of the alimentary system.
Female Reproductive System
Porcine Reproductive and Respiratory Syndrome (PRRS Virus, Enveloped RNA Virus): The pathogenesis of the initiating phases of PRRS is discussed in the section on the Respiratory System, Mediastinum, and Pleurae. Although unknown, the mechanism and type of injury that occurs in the lung also probably affects a wide variety of cells in the placenta, fetal membranes, and fetus. Injury can be observed in fetal myocytes; however, it is unclear as to whether loss of myocytes is attributable to necrosis, apoptosis, or atrophy. Gross lesions include abortions (born weak) and fetal deaths (mummification, stillbirths). Virus probably spreads to the placenta in virus-infected macrophages within the circulatory system via leukocytic trafficking from an initial site of virus replication in another body system such as the lung or uterus. It is likely that virus-infected macrophages transfer virus to fetal macrophage-like cells in the placentome, which then spread virus to all organ systems in the fetus. Although all fetuses in a litter may not be infected, it has been shown that pig fetuses in all stages of gestation can be infected with and support replication of virus resulting in normal, born weak, stillborn, and mummified fetuses.
Bovine Herpesvirus Abortion (Bovine Herpesvirus 1: Alphaherpesvirus, Enveloped DNA Virus): See infectious bovine rhinotracheitis in the section on the Respiratory System, Mediastinum, and Pleurae. Gross lesions include abortions (born weak) and fetal deaths (mummification, stillbirths). Virus-infected mucosal macrophages, lymphocytes, or dendritic cells migrate in lymphatic vessels via leukocyte trafficking and spread virus to regional lymph nodes such as the tracheobronchial. Here, it infects macrophages and lymphocytes, which spread it to the circulatory system and placenta via cell-free viremia or leukocyte trafficking through postcapillary venules or lymphatic vessels and the thoracic duct. It is not clear how virus spreads from the uterus, to the placenta, and then to the fetus, but some form of a fetal macrophage-like cell probably intervenes in the placentome. Cell types and injury involved are undetermined.
Equine Herpesvirus Abortion (Equine Herpesvirus 1 and 4: Alphaherpesvirus, Enveloped DNA Virus): See equine viral rhinopneumonitis in the section on the Respiratory System, Mediastinum, and Pleurae. Gross lesions include abortions (born weak) and fetal deaths (mummification, stillbirths). Virus-infected mucosal macrophages, lymphocytes, or dendritic cells migrate in lymphatic vessels via leukocyte trafficking and spread virus to regional lymph nodes such as the tracheobronchial. Here, it infects macrophages and lymphocytes, which spread it to the circulatory system and placenta via cell-free viremia or leukocyte trafficking through postcapillary venules or lymphatic vessels and the thoracic duct. It is not clear how virus spreads from the uterus, to the placenta, and then to the fetus, but some form of a fetal macrophage-like cell probably intervenes in the placentome. Cell types and injury involved are undetermined.
Infectious Pustular Vulvovaginitis/Balanoposthitis (Bovine Herpesvirus 1: Alphaherpesvirus, Enveloped DNA Virus): See infectious bovine rhinotracheitis in the section on the Respiratory System, Mediastinum, and Pleurae. Gross lesions include erosion and ulcerations with hemorrhage of reproductive mucosae (see Fig. 18-46). Virus-infected mucosal macrophages, lymphocytes, or dendritic cells migrate in lymphatic vessels via leukocyte trafficking and spread virus to regional lymph nodes such as the tracheobronchial. Here, it infects macrophages and lymphocytes, which spread it to the circulatory system and placenta via cell-free viremia or leukocyte trafficking through postcapillary venules or lymphatic vessels and the thoracic duct. Virus then spreads to epithelial cells of the mucous membranes of the penis, prepuce, vulva, or vagina via cell-free viremia or leukocyte trafficking. Because virus causes death of infected cells and thus erosions and ulcerations of mucosae, it may also be spread via direct contact (venereal disease) of virus-infected mucosae from the penis or prepuce with mucosae of the vulva or vagina, or vice-versa, during breeding.
Coital Exanthema (Equine Herpesvirus 3: Alphaherpesvirus, Enveloped DNA Virus): The mechanism of injury in coital exanthema is dysfunction and death of mucosal epithelial cells of the male and female reproductive systems. Gross lesions include active hyperemia, hemorrhage, erosions, and ulceration of affected mucosae, leading to an acute inflammatory response (see Fig. 18-48). Horses encounter virus through direct contact (venereal disease) with infected horses during breeding. Mucosae do not need to be injured for virus to infect cells. Virus can also spread mechanically via hands, gloves, instruments, palpation sleeves, and sponges, if contaminated with virus. Although unidentified, equine herpesvirus 3 probably expresses attachment proteins in its envelope that attach and bind to specific receptors on cells of reproductive mucosae.
Equine Viral Arteritis (Arterivirus, Enveloped RNA Virus): See equine viral arteritis in the section on the Cardiovascular System and Lymphatic Vessels. Gross lesions include abortions (born weak) and fetal deaths (mummification, stillbirths). Virus-infected macrophages spread virus to mucosal lymphocytes and macrophages of the endometrium and then to blood vessels of the endometrium leading to endothelial cell and myocyte necrosis and a necrotizing vasculitis. Trophoblasts can also be infected with virus. Abortion probably results from placenta-endometrial dysfunction (likely vascular necrosis and vasculitis), potentially resulting in detachment. The migration of virus in the placenta or the fetus and the cell types and injury involved are undetermined, but some form of a fetal macrophage-like cell is probably involved. Stallion semen is also a likely source of virus (accessory sex glands). It is deposited on mucosae and virus likely infects and replicates in mucosal macrophages and as they migrate through the mucosae and then is spread by these cells locally through leukocyte trafficking to submucosae where they infect and replicate in tissue macrophages and lymphocytes. These cells then migrate to blood vessels and injure endothelial cells of the endometrium as described previously.
Porcine Parvovirus Abortion (Parvovirus, Nonenveloped DNA Virus): See canine parvovirus enteritis and parvovirus-induced cerebellar hypoplasia in the sections on the Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity and the Nervous System, respectively. The mechanism of injury is dysfunction and potentially death of placental and fetal cells. Gross lesions include reproductive failure, embryonic death, fetal resorption, stillbirths, and mummified fetuses (see Fig. 18-44).
Pigs encounter virus through direct contact with fomites from fluids or tissues of the reproductive system, placenta, or aborted fetuses. Virus can also be transferred mechanically via hands, gloves, and instruments, if they are contaminated with virus-infected body fluids. It is ingested or inhaled and deposited on mucosae of the oral, nasal, and pharyngeal cavities, especially of the tonsil. It has not been determined if and how virus penetrates the mucus layer to gain access to tonsillar mucosal epithelial cells. Virus likely infects and replicates in mucosal macrophages and dendritic cells as they migrate through the mucus layer and mucosae and then is spread by these cells locally through leukocyte trafficking to the submucosa where they infect and replicate in tissue macrophages, lymphocytes, and dendritic cells of the tonsil (MALT). A cell-free viremia may also occur. These cells spread virus in lymphatic vessels via leukocyte trafficking to regional lymph nodes where they infect and replicate in similar cells, and spread virus to the circulatory system and systemically to lymph nodes through postcapillary venules or lymphatic vessels and the thoracic duct. From the circulatory system, it is unclear how virus spreads from the uterus, to the placenta, and then to the fetus; however, studies suggest the spread of virus to the fetus occurs via leukocyte trafficking of placental fetal macrophage-like cells. Although unidentified, porcine parvovirus probably has capsid attachment proteins that bind to glycosylated cell membrane receptors (likely sialic acid-bearing cell surface receptors) of host target cells in the uterus, placenta, and fetus. Virus has been identified in placental and fetal endothelial cells and in most tissues of virus-infected fetuses. Parvoviruses only infect and replicate in dividing cells because they require a host cell derived duplex transcription template (see Chapter 6), which is available when cells divide during S-phase of the cell cycle. Parvoviruses are unable to turn on DNA synthesis in host cells, so they must wait for host cells to enter the S-phase of the cell cycle before they can infect cells. It is likely that the high mitotic rate of developing and growing fetal tissues is conducive to infection by virus. Virus-induced death of cells likely causes injury resulting in embryonic death, fetal resorption, stillbirths, and mummified fetuses.
Ear and Eye
Feline Herpetic Keratitis (Feline Herpesvirus 1: Alphaherpesvirus, Enveloped DNA Virus): See feline viral rhinotracheitis, equine viral rhinopneumonitis, and infectious bovine rhinotracheitis in the section on the Respiratory System, Mediastinum, and Pleurae. The mechanism of injury is death of epithelial cells of the cornea. Gross lesions include corneal ulcerations; however, with severe injury involvement of the underlying corneal stromal can occur leading to edema, neovascularization, collagenization, and inflammation. These secondary lesions are attributable to inflammation and its mediators especially those derived from cytotoxic T lymphocytes. Cats encounter virus in fomites from body fluids, such as saliva and eye and nasal secretions, contaminated through direct contact with virus-infected cats. Virus is deposited on mucosae of conjunctiva where it infects and replicates in the epithelium. Viral envelope glycoproteins are used to attach to and enter these cells via glycosaminoglycan receptors on conjunctival epithelial cells. Virus replicates in mucosal epithelial cells and with cell death virus is released to spread in conjunctival fluids to corneal epithelial cells where the processes are repeated and the corneal epithelium becomes ulcerated and inflamed.